

International Journal of Pathology and Clinical Research
Histological Findings in the Kernicterus-Associated Vulnerable Brain Regions are Linked to Neurodegeneration, Alterations in Astrocyte and Pericyte Distribution, and Vascular Modifications
Inês Palmela1, Pedro Pereira2,3, Masaharu Hayashi4, Dora Brites1,5 and Maria A. Brito1,5*
1Research Institute for Medicines, Faculdade de Farmácia, Universidade de Lisboa, Portugal
2Laboratory of Neuropathology, Centro Hospitalar Lisboa Norte, Portugal
3Neuromuscular Unit, Institute of Molecular Medicine, Faculdade de Medicina, Universidade de Lisboa, Portugal
4Department of Brain Development and Neural Regeneration, Tokyo Metropolitan Institute of Medical Science, Japan
5Department of Biochemistry and Human Biology, Faculdade de Farmácia, Universidade de Lisboa, Portugal
*Corresponding author:
Maria A. Brito, Medicines Research Institute (iMed. ULisboa), Faculdade de Farmácia, Universidade de Lisboa, Avenida Professor Gama Pinto, 1649-003 Lisbon, Portugal, Tel: 351217946449, Fax: 351217946491, E-mail: abrito@ff.ulisboa.pt
Int J Pathol Clin Res, IJPCR-1-003, (Volume 1, Issue 1), Research Article; ISSN: 2469-5807
Received: April 14, 2015 | Accepted: April 27, 2015 | Published: April 29, 2015
Citation: Palmela I, Pereira P, Hayashi M, Brites D, Brito MA (2015) Histological Findings in the Kernicterus-Associated Vulnerable Brain Regions are Linked to Neurodegeneration, Alterations in Astrocyte and Pericyte Distribution, and Vascular Modifications. Int J Pathol Clin Res 1:003. 10.23937/2469-5807/1510003
Copyright: © 2015 Palmela I, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Kernicterus is a severe manifestation of neonatal unconjugated hyperbilirubinemia. We investigated the neuro-glia-vascular alterations in autopsy material from three infants with kernicterus. Histological and immunohistochemical studies were performed in the cerebellum, hippocampus and basal ganglia, the most vulnerable brain regions to bilirubin-induced neurotoxicity. The data obtained were compared with the relatively spared temporal cortex, as well as with three aged-matched controls with no hyperbilirubinaemia. Our data showed a reduction of the external germinal layer thickness e kernicterus cases cerebellum, indicating that bilirubin compromises the neural progenitor cells. Results also showed that neuronal dysfunction, including neuronal death and reduced neuronal bodies, was prevalent in the cerebellum, hippocampus and basal ganglia. The hippocampus was the region presenting the greatest neuronal loss and vacuolation, also showing astrogliosis and loss of pericyte vascular coverage. A marked decrease in the basement membrane collagen IV immunoreactivity was observed in the cerebellum, a region presenting increased vessel density, particularly in the cerebellar cortex. Moreover, based on the enhanced caveolin-1 expression observed in the cerebellum and hippocampus we hypothesize that a transcellular hyperpermeability may have been involved in cases of kernicterus. The temporal cortex did not show signs of endothelial dysfunction and was the one with the lowest microvessel density and the highest basement membrane thickness, features that may account to the limited bilirubin passage across the blood-brain barrier into the brain and to the low propensity of the temporal cortex to kernicterus.
Conclusion: The results obtained in three post-mortem brain samples of children with kernicterus and comorbid factors indicate that neuronal impairment and astrocytosis occur in parallel with microvascular alterations commonly associated with blood-brain barrier impairment.
Keywords
Astrogliosis, Basement membrane, Blood-brain barrier, Caveolae, Hyperpermeability, Kernicterus, Pericyte vascular coverage, Transcytosis, Unconjugated bilirubin, Vascularization
Introduction
Neonatal jaundice is extremely common in the first week of life, affecting 60 to 85% of neonates [1]. The condition is usually benign and resolved with no treatment requirement. However, under circumstances such as prematurity and glucose-6-phosphate dehydrogenase deficiency [2,3] unconjugated bilirubin (UCB) levels may increase dramatically or extend beyond the first week of life and lead to acute bilirubin encephalopathy, or kernicterus, a potentially lethal disease [1,4,5]. Bilirubin entry into the brain is facilitated by drugs that displace bilirubin from its albumin binding site, by reduced albumin binding capacity to bilirubin, but also by an increased brain blood flow and enhanced permeability of the blood-brain barrier (BBB) [6,7]. Recently, ex vivo studies of a kernicterus case revealed angiogenic sprouting and the presence of blood-borne components in the brain parenchyma, together with neuronal impairment [8,9]. In addition, in vitro studies in conditions mimicking a moderate and severe neonatal jaundice (UCB/albumin molar ratios of 0.5 and 1.0, respectively), revealed that UCB induces the disruption of tight junctions and increases caveolae formation, reflecting an enhanced paracellular and transcellular hyperpermeability, respectively [10,11]. Moreover, it was recently demonstrated that UCB also compromises pericytes [12], which are known to play a key role in the maintenance of BBB properties [13].
Kernicterus is characterized by a preferential deposition of UCB in specific brain regions, such as the cerebellum, hippocampus, and basal ganglia, as well as within the cytoplasm of macrophages and neurons [8,14,15]. Several reports of autopsy cases of kernicterus have shown extensive neuronal loss, myelination defects and oxidative stress in these three brain areas [9,14-17]. Numerous studies with animal models of severe hyperbilirubinemia have demonstrated cerebellar hypoplasia and significant loss of Purkinje and granule neurons [18-20]. In addition, microglia activation and impaired long-term synaptic plasticity were observed in in vitro and ex vivo studies using hippocampus [21,22]. When evaluating the response of neurons isolated from such regions to UCB cytotoxicity, the hippocampal neurons were the most susceptible [23] with a compromised neuronal differentiation, development, and plasticity [24]. However, and despite the well-known region-specific vulnerability to UCB harmful effects, the underlying reasons for such specific pattern are still unclear.
In the present study we investigated the histopathological alterations in the cerebellum, hippocampus and basal ganglia in post-mortem samples from kernicterus cases, and evaluated the associated neuronal-glial-vascular changes by comparing data with that from non-icteric controls. To better understand the region-specific pattern of kernicterus, we decided to also include the temporal cortex in the study, due to its assumed less vulnerability. Changes in the number of neurons (and cell body area), of astrocytes and of vessels, as well as in pericyte vascular coverage, collagen IV and caveolin-1 in vessel walls were assessed.
Materials and Methods
Subject description
In this work, we re-examined autopsy material from three neonates with diagnosed kernicterus and same number of matched controls already described [17]. In summary, the patients consisted in two males and one female that experienced hyperbilirubinemia during the neonatal period with the diagnosis of acute bilirubin encephalopathy (kernicterus) and associated morbidity made at autopsy. Kernicterus case 1 (K1), a female, was born at 29 weeks of gestation with 1.2kg birth weight. She presented mild asphyxia, and developed dyspnea and severe jaundice one day after birth, dying of respiratory failure due to pneumonia, at the 4th day of life. No therapy was performed for severe jaundice. Autopsy demonstrated intracerebral hemorrhage and severe pneumonia, in addition to kernicterus. Kernicterus case 2 (K2) was a male, born with 2.2kg birth weight, at 35 weeks, due to premature rupture of amnionic membrane, without asphyxia. The patient revealed severe jaundice and abdominal distention with frequent vomiting and received phototherapy. Seven days after birth, the patient developed fever and dyspenea, and died from respiratory failure at the 10th day of life. Autopsy demonstrated suppurative bacterial meningitis, gastric ulcer and liver abscess with severe jaundice, in addition to kernicterus. Kernicterus case 3 (K3) was a male, born with 3.5kg, at 39 weeks, with mild asphyxia and developed jaundice three days after birth. The infant received phototherapy and revealed poor sucking reflex and suffered a brief convulsion at the 4th day of life. Five days after birth, he was admitted to the hospital with consciousness disturbance, dehydration and hypotonia. The patient died on the 11th day of life of subarachnoid and intraventricular hemorrhage. Autopsy demonstrated brain hemorrhage, kernicterus, brain oedema and systemic jaundice. Values for total bilirubin levels in these three cases were not made available and a deficiency in glucose-6-phosphate dehydrogenase enzymatic activity was not reported [17].
The control cases consisted in three males that died without any pathological changes in the central nervous system (CNS). Control case 1 (C1) was a male born with 2.9Kg, at 36 weeks/5days of gestation after toxemia and threatened abortion. The patient was healthy and had good sucking. Two days after birth, he developed hypoglycaemia that was improved by 10% glucose infusion. The patient died at the 4th day of life of sudden cardiorespiratory arrest. Autopsy demonstrated no changes in either the brain or general organs, and he was diagnosed as having sudden infant death syndrome. Control case 2 (C2) was a male born with 3.0Kg, at 40 weeks/3 days, with normal sucking. On the 3rd day of life, the patient died of sudden cardiorespiratory arrest. Autopsy demonstrated no changes in either the brain or general organs, and he was diagnosed as having sudden infant death syndrome. Control case 3 (C3) was a male, with 2.7Kg that died at 41 weeks of sudden intrauterine cardiac arrest just before birth. There was no abnormality in the baby blood tests, but the infection of group B Streptococcus was confirmed in the amniotic fluid. Autopsy demonstrated no changes in the brain, but revealed severe chorioamniotis of the placenta, vasculitis, and bronchopneumonia. Sepsis was considered an associated cause of death.
Tissue was obtained and used in a manner compliant with the Declaration of Helsinki, as revised in 1983. The family of each infant provided informed consent for neuropathological analysis. Brains of all subjects were fixed in buffered formalin solution for 2 weeks. Each formalin-fixed paraffin-embedded sample was cut coronally in 6μm sections as previously described. Except for some non-available sections (Supplementary Table 1,2), this study relied on analysis of the cerebellum, hippocampus and basal ganglia of the three kernicterus and three control cases.
Histology and immunohistochemistry
Neuronal-astrocyte-vascular parameters were evaluated in four brain regions: cerebelllum, hippocampus, basal ganglia and temporal cortex. Standard Hematoxylin-Eosin staining was performed for the histological evaluation of the different brain regions. For immunohistochemical analysis, a 3% H2O2 solution was used to inhibit endogenous peroxidase and antigen recovery was achieved by treatment with citrate buffer. Sections were incubated overnight at 4°C with the primary antibodies: mouse anti-cluster of differentiation 34 (CD34) diluted 1/100 (Leica Biosystems, # NCL-L-END), mouse anti-neurofilament diluted 1/40 (Invitrogen, #180171Z), rabbit anti-caveolin-1 diluted 1/100 (Cell Signaling, #3238S), rabbit anti-glial fibrillary acidic protein (GFAP) diluted 1/250 (Sigma-Aldrich, # G9269), mouse anti-μ-smooth muscle actin diluted 1/100 (AbDSerotec, #MCA2903Z) and mouse anti-collagen IV diluted 1/100 (Sigma-Aldrich, #C1926). Sections were incubated with EnVision detection system for 1 h at room temperature, followed by the development with 3,3-diaminobenzidine tetrahydrochloride and H2O2 solution, and finally counterstained with Mayer's hematoxylin. Images were acquired with a bright field microscope (Zeiss, model AxioSkop HBO50) with an integrated digital camera (Leica, model DFC490).
Data collection and statistical analysis
Data analysis was performed using the ImageJ software (National Institutes of Health, USA). Neuronal-astrocyte-vascular changes were analysed in a minimum of 15 independent photos per sample. Unless otherwise specified, analysis was performed in the overall cerebellum, hippocampus and basal ganglia. Cerebellar layer thickness was calculated by manually tracing a perpendicular line extending from the beginning to the end of each identifiable layer and measuring the respective line length. Analysis relied on the count of neuronal number, which was expressed per microscopic field, as well as on the determination of neuronal area, which was calculated by tracing the cell body with the referred software and expressed in μm2. The number of astrocytes was evaluated by counting the cells expressing the glial marker GFAP, while vascular features were assessed by the following markers: caveolin-1, α-smooth muscle actin and collagen IV. For the evaluation of vessel density, the number of vessels per field was counted and expressed as number of vessels per mm2 of brain tissue. The intensity and area of the exterior and interior (lumen) limits of the individual vessels were measured, and then the interior lumen data were deducted from the exterior lumen values in 8-bit pictures. All immunostaining intensities were normalized by vessel wall area, and expressed as intensity per μm2 of vessel wall. In the case of collagen IV it was evaluated the intensity of the immunostaining per μm2 of vessel wall, as well as the area (μm2) occupied by the component. Assessment of pericyte vascular coverage was based on the staining of α-smooth muscle actin, a known pericyte marker [13], surrounding the vessels. The total intensity of collagen IV, which was proportional to the pericyte vascular coverage, was measured and then normalized to the vessel caliber. Collagen IV was measured on transversely-sectioned vessels, while caveolin-1 and α-smooth muscle actin were evaluated on longitudinal vessels. Caliber of blood vessels was separated into three categories: under 7.5μm (small vessels, or capillaries), between 7.5 and 15μm (intermediate vessels), and between 15 and 30μm (large vessels). We only considered small and intermediate vessels directly related with the BBB-brain microvasculature, and, unless stated, data are a combination of such categories.
To ascertain that alterations in the kernicterus cases were due to hyperbilirubinemia and UCB deposition in specific brain regions, related to the diagnosis of kernicterus at autopsy, and not biased by the K1 prematurity, results were separately analysed and are shown in supplementary tables 1 and 2. Since the values obtained fitted within those of K2 and K3, data are expressed as means ± SEM. Significant differences between groups were determined by the two-tailed t-test performed on the basis of equal and unequal variance as appropriate. Statistical significance was considered when P values were lower than 0.05.
![]()
Supplementary Table 1: Neuronal density and neuron body area of each of the kernicterus (K) cases
View Supplementary Table 1
![]()
Supplementary Table 2: Astrocytic, perivascular and vascular data obtained for each of the kernicterus (K) cases
View Supplementary Table 2
Results
Histological findings in the kernicterus-associated vulnerable brain regions comprise degenerating neurons, parenchymal spongiosis/vacuolation and depletion of neurons
We started the histological examinations by observing the sections stained by Hematoxylin-Eosin (Figure 1). Common features observed in the cerebellum, hippocampus and basal ganglia of the samples collected from the three children with kernicterus included the eosinophilia of neurons, a characteristic of cells that are degenerating. Such neurons were predominantly observed in the hippocampus, but were also evident in the cerebellum and basal ganglia, but not in the temporal cortex (Figure 1a). Other common effects were an increased spongiosis (vacuolation) in the brain parenchyma, usually linked to the depletion of neurons, more evident in the hippocampus and also not noticed in the temporal cortex (Figure 1b). Injury may result from the cytoplasmic accumulation of bilirubin once it was not observed in the temporal cortex region, which is not susceptible to kernicterus, or in the matched controls (Figure 1c).
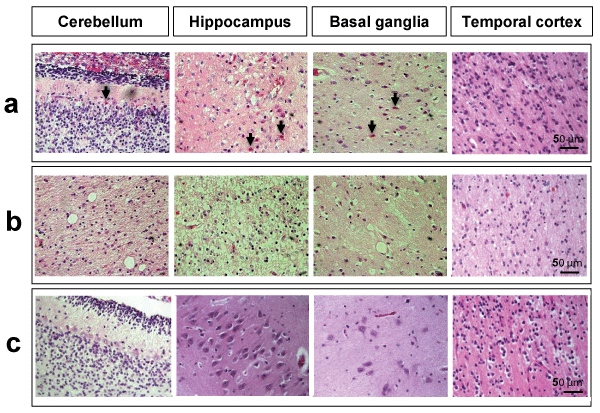
.
Figure 1: Histologic features of the brain parenchyma of kernicterus and control cases. Hematoxylin-Eosin staining revealed: (a) eosinophilic neurons (arrows) in the cerebellum, hippocampus and basal ganglia, but not in the temporal cortex of kernicterus cases; (b) spongiosis and vacuolation in the cerebellum, hippocampus and basal ganglia, but not in the temporal cortex of kernicterus cases; (c) no eosinophilic staining nor spongiosis or vacuolation in the control cases in any of the studied regions. Representative photos are shown.
View Figure 1
Since the loss of neurons is a kernicterus hallmark we next performed neurofilaments immunostaining and determined the thickness of cerebellar layers as well as the neuronal density and neuron body area (Figure 2). Bilirubin seems to be in the origin of the significant 34% reduction in the extent of the external germinal layer of the cerebellar cortex, not causing changes at the molecular layer, Purkinje cell layer and internal granular layer, shown in figure 2a. Interestingly, a reduction in the number of neurons per field was noticed in the Purkinje neurons of the cerebellar cortex (24%, Figure 2b), but even more obviously in the dentate nucleus neurons (65%, Figure 2c) when compared to control samples. Also to point out, cerebellar neurons did not reveal any alteration in the cell body dimension. In accordance with the histopathological findings depicted in figure 1 neuronal loss was particularly manifest in the third layer of the Ammon's horn (CA3, hippocampus) (Figure 2d). Besides the 73% decrease in neuronal density, cells were also changed in their cell body dimensions, revealing a 51% decrease in samples from the children that developed kernicterus. A very important 62% reduction of neuronal density and 39% in cell body dimension was similarly observed in the globus pallidus region of the basal ganglia (Figure 2e), when compared to matched controls.
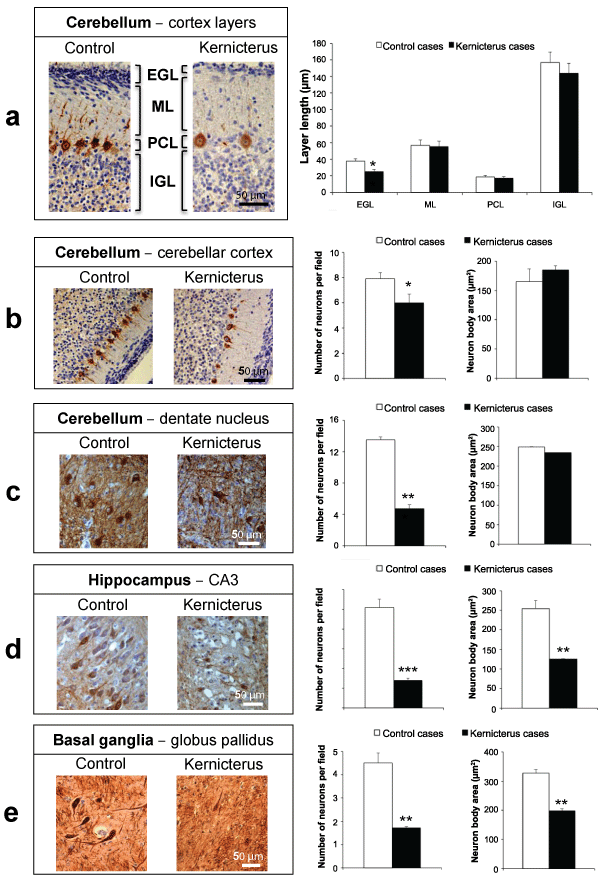
.
Figure 2: Neuronal alterations in the brain parenchyma of kernicterus cases.Immunostaining for the neuronal marker, neurofilaments, revealed: (a) a reduction in the thickness of the external germinal layer (EGL), with no alterations in those of the molecular layer (ML), Purkinje cell layer (PCL) and internal granular layer (IGL) of the cerebellar cortex; (b) a reduction in the number of Purkinje neurons in the cerebellar cortex, with no changes in their cell body area; (c) a reduction in the number of neurons in the dentate nucleus of the cerebellum, with no changes in their cell body area; (d) a reduction in both the number and cell body area of the neurons present in the third layer of the Ammon's horn (CA3) of the hippocampus; (e) a reduction in both the number and cell body area of the neurons present in the globuspallidus region of the basal ganglia. Representative photos are shown. *P< 0.05, **P< 0.01 and ***P< 0.001 vs. control cases.
View Figure 2
Analysis of the non-icteric brain established the specific neuronal, astroglial and vascular profiles of different brain regions (Table 1). The maximal neuronal density was typically manifest in CA3 hippocampus, with an approximate 5-fold increase over the other two evaluated brain regions (P< 0.05), which may have accounted for the increased neurotoxicity associated to kernicterus in this specific brain region. In addition, the larger neuron body area, mainly for cells located in the globuspallidus region when compared to the Purkinje neurons of the cerebellar cortex (P< 0.05), may justify the particular neuronal shrinkage in the hippocampus and basal ganglia observed in the kernicterus samples.
![]()
Table 1: Neuronal, astroglial and vascular profiles of different brain regions in non-icteric brain
View Table 1
Kernicterus-associated hippocampus presents alterations in the distribution of astrocytes and pericytes whereas cerebellum shows a decrease in the collagen IV component of the basement membrane
Astrogliosis was shown to occur in experimental models of hyperbilirubinemia [25-27], and in kernicterus cases [28]. Therefore, we anticipated to also find an increased number of astrocytes in the post-mortem samples from the cases of kernicterus. For that, we examined the density of GFAP labelled astrocytes. In the children that developed bilirubin encephalopathy astrogliosis was only significantly enhanced in the hippocampus (76%, Figure 3a), i.e. the region with major neuronal loss and vacuolation. On the other hand, it is interesting to point out that the region presenting the highest astrocytic density in control cases is the temporal cortex (Table 1), which is known for the lower susceptibility to UCB deposition, as mentioned above.
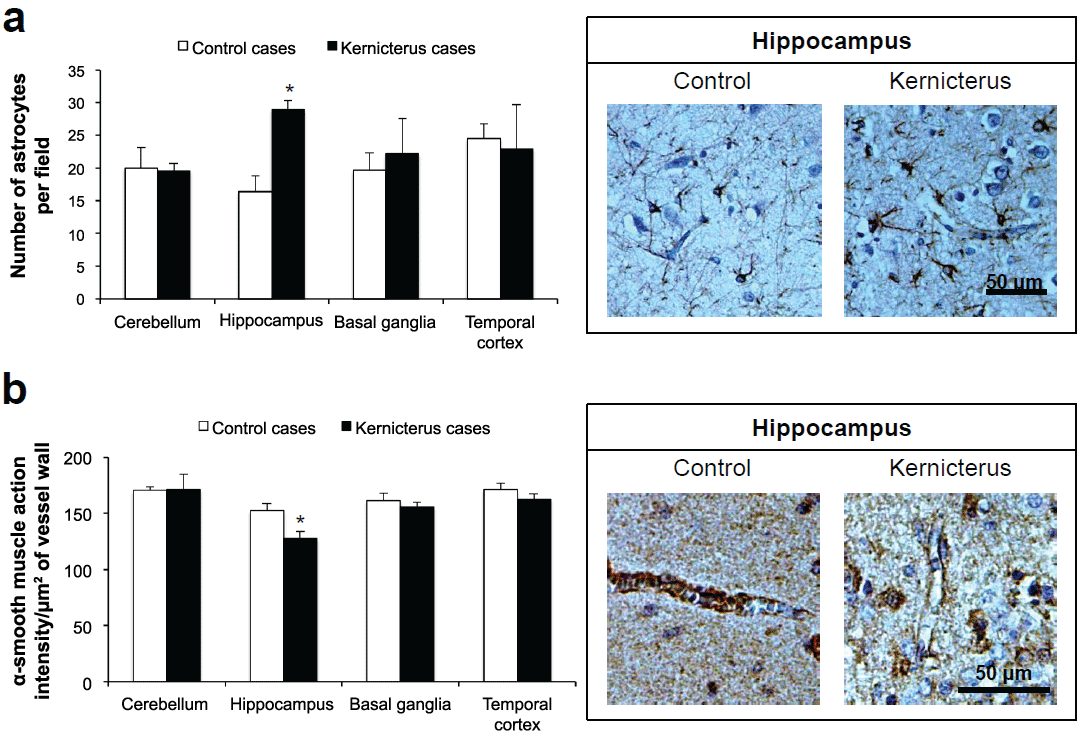
.
Figure 3: Changes in astrocyte number and pericyte vascular coverage in the brain parenchyma of kernicterus cases. (a) Immunostaining for the astrocyte marker, glial fibrillary acidic protein, showed an elevation in the number of these glial cells in the kernicteric hippocampus; (b) Immunostaining for the pericyte marker, α-smooth muscle actin, revealed a reduction in the pericyte vascular coverage in the hippocampus. Representative photos are shown. *P< 0.05 vs. control cases.
View Figure 3
The CNS microvascular pericytes are considered unique in promoting tissue survival [29] and the loss of pericyte vascular coverage has been associated to BBB impairment and neurodegeneration [30,31], namely in our own studies (Janota et al. submitted). Although pericytes are relatively quiescent in normal conditions, the picture changes during stress and injury, where the loss of pericytes may compromise the pericyte-astrocyte communication and contribute to a focal increase of BBB permeability. Thus, we next evaluated the pericyte vascular coverage by immunostaining of α-smooth muscle actin, a known pericyte marker [13]. It was interesting to verify that a 16% lack of pericyte vascular coverage was precisely located in the hippocampus (Figure 3b), the region that showed astrogliosis. In contrast, no differences were noted for the other two kernicterus-associated brain regions, nor for the temporal cortex or among brain regions in the matched controls.
The CNS pericyte is surrounded by the basal lamina on all sides. The basal lamina has been shown to become thicker or thinner in response to stress stimuli, and pericytes are known producers of type IV collagen among other basal lamina constituents [32]. To evaluate if the reduced coverage of microvascular pericytes or dysfunctional pericytes were responsible for a compromised basal lamina formation, we determined the collagen IV immunostaining in kernicterus samples and controls. A significant 31% decrease in the collagen IV immunoreactivity per vessel wall area was noticed in the cerebellum for both small and intermediate vessels (Figure 4a,4b), but not in the other regions. Thus, it is conceivable that bilirubin may have accounted to an increased fragility of these cerebellar pericytes. In addition, the 41% increase in the collagen area in the vessels of the temporal cortex, but not in all the other regions (Figure 4c,4d), may derive from defensive/adaptive mechanisms that this region possibly develops against bilirubin-induced injury.
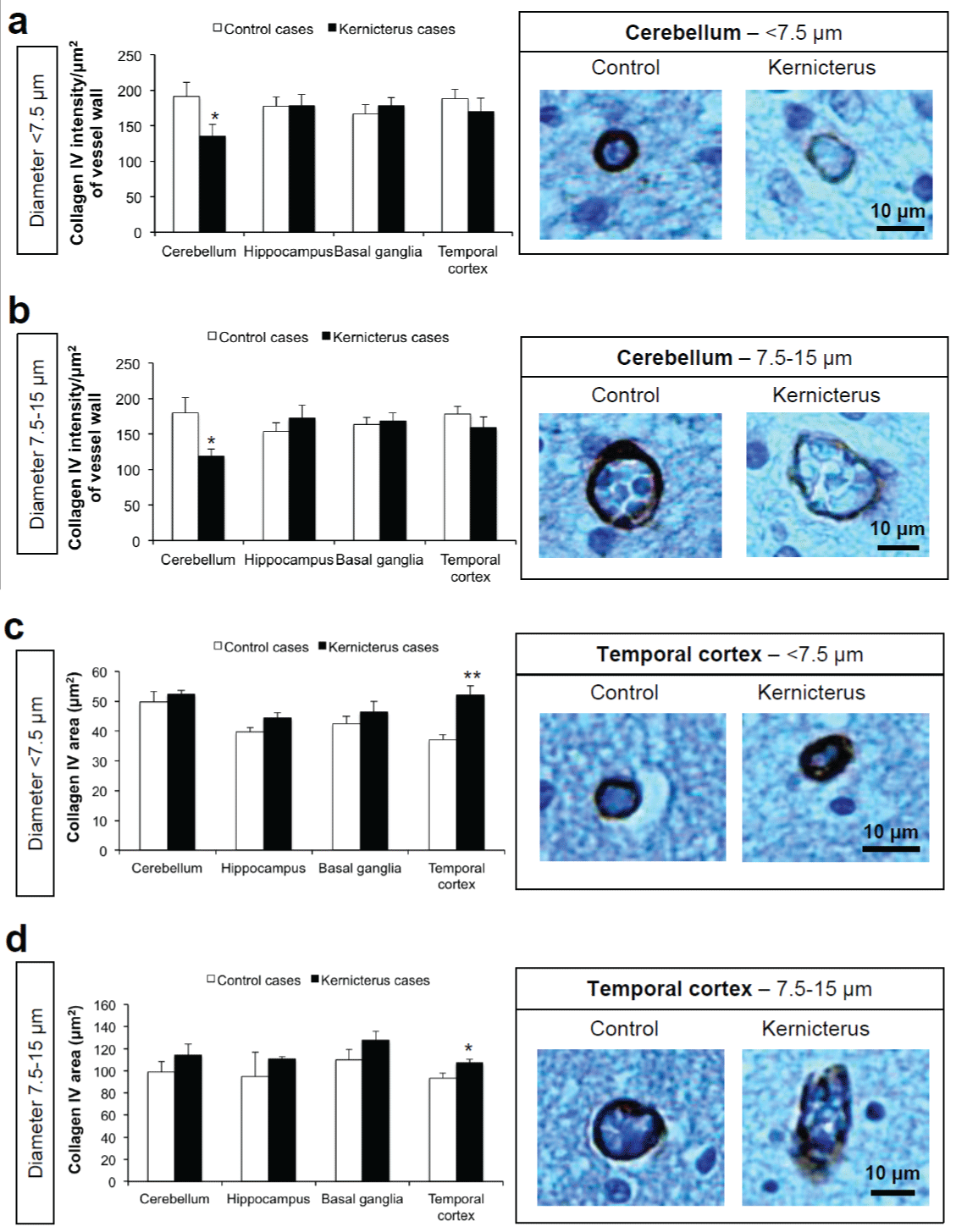
.
Figure 4: Changes in basement membrane assembly in the brain parenchyma of kernicterus cases. Immunostaining for the basement membrane component collagen IV showed: (a,b) a decreased intensity in small (< 7.5μm) and intermediate (7.5-15μm) vessels in the cerebellum; (c,d) an increased collagen IV area in small (< 7.5μm) and intermediate (7.5-15μm) vessels in the temporal cortex. Representative photos are shown. *P< 0.05 vs. control cases.
View Figure 4
Changes to blood vessels in the kernicterus brain
Bilirubin may disturb the membrane structure of the capillary endothelial cells [10,33]. Accordingly, vasogenicoedema and leakage of erythrocytes into the extravascular space were observed by Hematoxylin-Eosin staining in all the kernicterus-associated vulnerable brain regions (Figure 5a,5b). Additionally, analysis of the endothelial marker CD34 [34] revealed an increased vascularization of the kernicterus over control samples in the cerebellum (combined analysis of the cerebellar cortex and dentate nucleus, P< 0.05, Figure 5c), mainly due to the 28% increase in the number of vessels counted in the cerebellar cortex, since no difference was noticed in the dentate nucleus. Such modification may have contributed to the decrease of the basement membrane previously indicated in this region. In contrast, a 14% reduction in the number of vessels was evident in the globuspallidus (Figure 5c). However, since the number of neurons dramatically decreased in both regions of kernicterus cases, the ratio between the number of vessels/number of neurons per mm2 was actually increased from 0.8 to 2.3 (3-fold elevation, P< 0.01) in the dentate nucleus of the cerebellum and from 1.6 to 3.5 (2.2-fold, P< 0.01) in the globuspallidus of the basal ganglia.
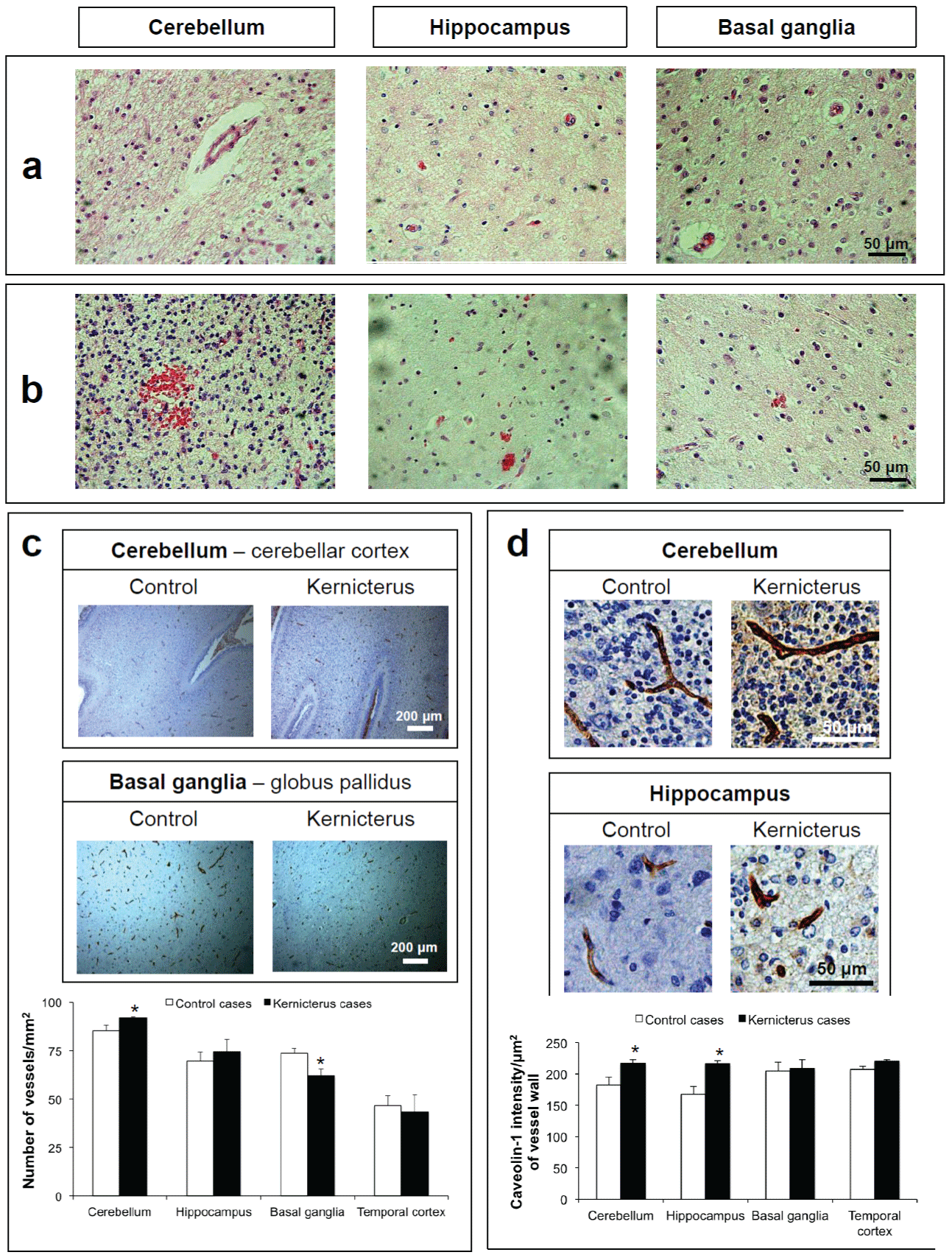
.
Figure 5: Blood vessels alterations in the brain parenchyma of kernicterus cases. Hematoxylin�Eosin staining revealed: (a) vascular oedema and (b) erythrocyte leakage. (c) Immunostaining for the endothelial marker, CD34, showed an increased number of vessels in the cerebellum (cerebellar cortex and dentate nucleus), while the basal ganglia presented a reduction in the number of microvessels per area, particularly in the globuspallidus. (d) Immunostaining for the caveolae marker, caveolin-1, revealed that the microvessels in the cerebellum and hippocampus present an elevated expression of the protein. Representative photos are shown. *P< 0.05 vs. control cases.
View Figure 5
Endothelial dysfunction was also evaluated based on caveolin-1 immunostaining, since its increase precedes barrier compromise [10] and is often observed in pathologies involving BBB breakdown [35]. Interestingly, our results showed an increased caveolin-1 immunoreactivity of 19 and 29% (P< 0.05) in the cerebellum and hippocampus (Figure 5d), respectively. Differences between brain regions in controls are indicated in table 1, where the temporal cortex revealed the lowest vessel density together with high astrocyte number and vessel intensity. These specific features are important when considering that cortex is not affected by kernicterus [5].
Discussion
Here, we report novel histopathological features observed in three cases of kernicteus during neonatal life. Although presenting associated risk factors, the kernicterus cases had hyperbilirubinemia as a common denominator, suggesting that UCB has been the dominant neurotoxic factor although it cannot be excluded the existence of concomitant aggravating conditions. The fact that the results were cross-compared with controls without hyperbilirubinemia or alterations of the CNS, and that infection was present in two of the kernicterus cases and one of the controls, further points to the relevance of UCB in the achieved neurovascular alterations. Moreover, since the results obtained for K1 (with 29 weeks gestation) did not differ from those for K2 and K3 (35 and 39 weeks gestation, respectively) suggests that prematurity was not the determinant of such changes. Nevertheless, additional human kernicterus cases should be studied to more firmly establish the relevance of the neuronal, astroglial and microvascular alterations here reported.
Our histological analysis revealed the existence of several neurons in the three brain regions (cerebellum, hippocampus and basal ganglia) with an orange cytoplasm, consistent with eosinophilia, which is a characteristic of neuronal degeneration [36]. High susceptibility of neurons to bilirubin and death was reported in several studies of kernicterus cases, especially in the cerebellum and hippocampus [8,9,14,15], and in animal models of severe hyperbilirubinemia [18,37]. Accordingly, we observed several other signs of neuronal compromise, such as parenchymal spongiosis and reduction in neuronal density and body area, together with a decreased layer thickness of the cerebellar cortex. Such findings seem to be derived from the significant diminution in the external germinal layer, a layer constituted by a stem cell population that disappears during development as the cells migrate to the internal granular layer and differentiate into granule cells [38]. Similar observation was noticed in a mouse model of severe jaundice [18], and in other injurious conditions [39,40], probably due to a deficient granule cell development that may compromise the survival of Purkinje neurons [41]. Interestingly, Robert et al. [42] reported the existence of cell cycle perturbation and apoptosis related to high bilirubin concentrations in the cerebellar tissue, mainly in granular cells.
Brain parenchyma homeostasis depends on complex interactions of neurons with glial and vascular cells within the neurovascular unit (NVU) [13] and dysfunction of the BBB has been associated with the onset and progression of numerous pathologies [43,44]. In the present study it was observed a marked elevation of astrocytic density in the kernicteric hippocampus. We have previously indicated that astrocytes may trigger an early neuroprotection against UCB injury [45], while adverse secondary impacts later occur [46], explaining why irreversible brain damage usually develops after the first day of post-natal life. Upon exposure to UCB, the release of pro-inflammatory cytokines by astrocytes [26,47], endothelial cells [48] and pericytes [12] may increase microvascular permeability [49] and sustain the UCB-induced impairment of the BBB observed in in vitro studies [10,11].
The interaction of pericytes with brain endothelial cells is believed to improve barrier function [50] and reduced pericyte coverage increases endothelial permeability [51-54]. In addition, pericyte deficiency was shown to contribute to the fragility of specific brain areas during development [55]. Here, we report for the first time a reduced pericyte vascular coverage in the kernicteric hippocampus, which is in line with the loss of pericytes viability and enhanced apoptotic death that we recently observed in primary cultures of human brain pericytes exposed to UCB in conditions mimicking a moderate and severe unconjugated hyperbilirubinemia [12]. Therefore, the loss of pericytes and the poorer pericyte vascular coverage suggests a microvascular fragility and BBB disruption, probably accounting to the extravasation of albumin into the brain parenchyma observed in a premature infant with kernicterus [8]. Interestingly, the depletion of pericyte covering the vasculature in the hippocampus seems to be accompanied by the above discussed astrocytes activation, raising the question of whether the loss of pericytes results from astrocytes activation or is in the origin of the observed astrogliosis. It is worthwhile to point out that the loss of pericyte vascular coverage here observed based on the immunostaining of the pericyte marker α-smooth muscle actin is in line with that observed by others [30] and by ourselves (Janota et al. submitted) in Alzheimer's disease patients and animal models based on analysis of other pericyte markers, CD13 and platelet derived growth factor receptor-β, These observations reinforce the role of pericytes in the pathogenesis of central nervous system diseases and point to their modulation as a possible therapeutic approach.
Amongst the many interactions within the NVU, the direct contacts of endothelial cells, astrocytes, and pericytes with the basement membrane are crucial for proper barrier function [13,56]. Therefore, the clear reduction in collagen IV immunoreactivity in cerebellar microvessels of the kernicterus cases reinforce the assumption of vascular impairment, in agreement with other pathological conditions [57-60] and with the neovascularization-associated degradation of collagen IV [61], herewith also observed. On the other hand, the enhanced thickness of the basement membrane observed in the temporal cortex suggests that this region is less permeable to UCB, compatible with the absence of yellow staining in this brain region in kernicterus cases compatible with no UCB deposition [8,62].
Analysis of the brain parenchyma also revealed signs of vascular oedema and the presence of erythrocytes in the cerebellum, hippocampus and basal ganglia, but not in the temporal cortex. Such events are in line with the microvascular fragility and hyperpermeability suggested by the diminished pericyte vascular coverage and basement membrane compromise observed in kernicterus-associated regions. Such endothelial permeability was already noticed in another kernicterus case [9]. Here, an increase in the number of microvessels was evident in the cerebellum, and particularly in the cerebellar cortex. Although dentate nucleus region of the cerebellum did not present changes in this parameter, its number relatively to that of neurons was considerably enhanced. Contrary to the cerebellum, the basal ganglia presented a lower number of microvessels, but the neuronal density was far more reduced. Thus, we may assume a higher number of vessels per neuron, especially in globus pallidus and dentate nucleus. Moreover, the high number of vessels observed in controls in the brain regions most susceptible to kernicterus, may determine such areas as preferential targets for UCB injury.
Caveolin-1 is the major structural protein of caveolae that are responsible for the transcellular transport of macromolecules such as albumin. Elevated levels have been directly associated with the reduction of intercellular junction proteins [63-65] and consequently, increased endothelial permeability and BBB breakdown [66-69]. The elevation of caveolin-1 immunoreactivity in the cerebellum and hippocampus microvessels, consistent with the increased number of caveolae and caveolin-1 previously found in human brain microvascular endothelial cells exposed to UCB [10], may therefore account to the entrance of albumin-bound UCB into the brain parenchyma.
Taken collectively, the results of this study provide new insights into the neuropathology of kernicterus and to the pattern of UCB deposition in specific brain regions, such as the cerebellum, the hippocampus and the basal ganglia. Specific vascular characteristics of the areas affected and not affected may preside to the selective damage observed in kernicterus. Our data corroborate that neuronal dysfunction, including neuronal loss and reduced neuronal body, is particularly evident in the cerebellum, hippocampus and basal ganglia, the most vulnerable areas to kernicterus. In addition, the decreased layer thickness observed in the cerebellar cortex eventually may reflect a compromised maturation. Changes in astrocytic density, lower vascular pericyte coverage, reduced basement membrane and enhanced microvascularization seem to be responsible for the selective neuropathological damage in kernicterus. Another interesting observation relies on the increased ratio between the number of vessels/number of neurons per mm2 of brain tissue in the cerebellum and globuspallidus remaining to clarify whether it is determined by a decreased number of neurons or enhanced vascularization. Furthermore, the elevated expression of caveolin-1 facilitating the passage of albumin-bound UCB across the endothelium towards the brain, may subsequently contribute to escalate the severity of bilirubin encephalopathy. Also relevant are the findings obtained for the temporal cortex that did not present signs of endothelial dysfunction in kernicterus. Indeed, this is the region with the largest basement membrane area in the pathological condition, which shall difficult UCB permeation. On the other hand, the greatest astrocytic density and pericyte vascular coverage, together with the lowest microvessels density, points to a more functional BBB and suggests a reduced UCB load to this region in control conditions. The disruptive effects of immaturity and associated risk factors to the kernicterus condition still occurring in premature babies may include additional aspects to those here indicated. Collectively, the novel data herewith reported depict the involvement of vascular dysfunction in the neuronal compromise by neonatal kernicterus and provide novel insights for the selective regional pattern of bilirubin deposition in kernicterus.
Acknowledgments
This work was supported by FEDER (COMPETE Programme) and by National funds (FCT - Fundação para a Ciência e a Tecnologia): project PEst-OE/SAU/UI4013/2011 and 2012 to iMedULisboa, PTDC/SAU-FCF/68819/2006 (to M. A. B.), and a PhD fellowship (SFRH/BD/61646/2009) to I.P.
References
-
Brites D, Brito MA (2012) Bilirubin toxicity. In: Stevenson DK, Maisels MJ, Watchko JF Care of the Jaundiced Neonate. Mc-Graw-Hill Companies Inc 115-143.
-
Costa S, De Carolis MP, De Luca D, Savarese I, Romagnoli C (2008) Severe hyperbilirubinemia in a glucose-6-phosphate dehydrogenase-deficient preterm neonate: could prematurity be the main responsible factor? Fetal Diagn Ther 24: 440-443.
-
de Luca D, Virdis A, Pietro ML, Costa S, de Carolis MP, et al. (2008) Heterologous assisted reproduction and kernicterus: the unlucky coincidence reveals an ethical dilemma. J Matern Fetal Neonatal Med 21: 219-222.
-
American Academy of Pediatrics Subcommittee on Hyperbilirubinemia (2004) Management of hyperbilirubinemia in the newborn infant 35 or more weeks of gestation. Pediatrics 114: 297-316.
-
Shapiro SM (2010) Chronic bilirubin encephalopathy: diagnosis and outcome. Semin Fetal Neonatal Med 15: 157-163.
-
Hansen TW, Oyasaeter S, Stiris T, Bratlid D (1989) Effects of sulfisoxazole, hypercarbia, and hyperosmolality on entry of bilirubin and albumin into brain regions in young rats. Biol Neonate 56: 22-30.
-
Hansen TW (2001) Bilirubin brain toxicity. J Perinatol 21 Suppl 1: S48-51.
-
Brito MA, Pereira P, Barroso C, Aronica E, Brites D (2013) New autopsy findings in different brain regions of a preterm neonate with kernicterus: neurovascular alterations and up-regulation of efflux transporters. Pediatr Neurol 49: 431-438.
-
Brito MA, Zurolo E, Pereira P, Barroso C, Aronica E, et al. (2012) Cerebellar axon/myelin loss, angiogenic sprouting, and neuronal increase of vascular endothelial growth factor in a preterm infant with kernicterus. J Child Neurol 27: 615-624.
-
Palmela I, Sasaki H, Cardoso FL, Moutinho M, Kim KS, et al. (2012) Time-dependent dual effects of high levels of unconjugated bilirubin on the human blood-brain barrier lining. Front Cell Neurosci 6: 22.
-
Cardoso FL, Kittel A, Veszelka S, Palmela I, Tóth A, et al. (2012) Exposure to lipopolysaccharide and/or unconjugated bilirubin impair the integrity and function of brain microvascular endothelial cells. PLoS One 7: e35919.
-
Brito MA, Palmela I, Cardoso FL, Sá-Pereira I, Brites D (2014) Blood-brain barrier and bilirubin: clinical aspects and experimental data. Arch Med Res 45: 660-676.
-
Sá-Pereira I, Brites D, Brito MA (2012) Neurovascular unit: a focus on pericytes. Mol Neurobiol 45: 327-347.
-
Perlman JM, Rogers BB, Burns D (1997) Kernicteric findings at autopsy in two sick near term infants. Pediatrics 99: 612-615.
-
Zangen S, Kidron D, Gelbart T, Roy-Chowdhury N, Wang X, et al. (2009) Fatal kernicterus in a girl deficient in glucose-6-phosphate dehydrogenase: a paradigm of synergistic heterozygosity. J Pediatr 154: 616-619.
-
Ahdab-Barmada M, Moossy J (1984) The neuropathology of kernicterus in the premature neonate: diagnostic problems. J Neuropathol Exp Neurol 43: 45-56.
-
Hachiya Y, Hayashi M (2008) Bilirubin encephalopathy: a study of neuronal subpopulations and neurodegenerative mechanisms in 12 autopsy cases. Brain Dev 30: 269-278.
-
Bortolussi G, Zentilin L, Baj G, Giraudi P, Bellarosa C, et al. (2012) Rescue of bilirubin-induced neonatal lethality in a mouse model of Crigler-Najjar syndrome type I by AAV9-mediated gene transfer. FASEB J 26: 1052-1063.
-
Lin S, Wei X, Bales KR, Paul AB, Ma Z, et al. (2005) Minocycline blocks bilirubin neurotoxicity and prevents hyperbilirubinemia-induced cerebellar hypoplasia in the Gunn rat. Eur J Neurosci 22: 21-27.
-
Yamamura H, Takagishi Y (1993) Cerebellar hypoplasia in the hyperbilirubinemic Gunn rat: morphological aspects. Nagoya J Med Sci 55: 11-21.
-
Liaury K, Miyaoka T, Tsumori T, Furuya M, Wake R, et al. (2012) Morphological features of microglial cells in the hippocampal dentate gyrus of Gunn rat: a possible schizophrenia animal model. J Neuroinflammation 9: 56.
-
Chang FY, Lee CC, Huang CC, Hsu KS (2009) Unconjugated bilirubin exposure impairs hippocampal long-term synaptic plasticity. PLoS One 4: e5876.
-
Vaz AR, Silva SL, Barateiro A, Falcão AS, Fernandes A, et al. (2011) Selective vulnerability of rat brain regions to unconjugated bilirubin. Mol Cell Neurosci 48: 82-93.
-
Fernandes A, Falcão AS, Abranches E, Bekman E, Henrique D, et al. (2009) Bilirubin as a determinant for altered neurogenesis, neuritogenesis, and synaptogenesis. Dev Neurobiol 69: 568-582.
-
Barateiro A, Domingues HS, Fernandes A, Relvas JB, Brites D (2014) Rat cerebellar slice cultures exposed to bilirubin evidence reactive gliosis, excitotoxicity and impaired myelinogenesis that is prevented by AMPA and TNF-a inhibitors. Mol Neurobiol 49: 424-439.
-
Fernandes A, Silva RF, Falcão AS, Brito MA, Brites D (2004) Cytokine production, glutamate release and cell death in rat cultured astrocytes treated with unconjugated bilirubin and LPS. J Neuroimmunol 153: 64-75.
-
Yueh MF, Chen S, Nguyen N, Tukey RH (2014) Developmental onset of bilirubin-induced neurotoxicity involves Toll-like receptor 2-dependent signaling in humanized UDP-glucuronosyltransferase1 mice. J Biol Chem 289: 4699-4709.
-
Steinborn M, Seelos KC, Heuck A, von Voss H, Reiser M (1999) MR findings in a patient with Kernicterus. Eur Radiol 9: 1913-1915.
-
Bonkowski D, Katyshev V, Balabanov RD, Borisov A, Dore-Duffy P (2011) The CNS microvascular pericyte: pericyte-astrocyte crosstalk in the regulation of tissue survival. Fluids Barriers CNS 8: 8.
-
Sengillo JD, Winkler EA, Walker CT, Sullivan JS, Johnson M, et al. (2013) Deficiency in mural vascular cells coincides with blood-brain barrier disruption in Alzheimer's disease. Brain Pathol 23: 303-310.
-
Sagare AP, Bell RD, Zhao Z, Ma Q, Winkler EA, et al. (2013) Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat Commun 4: 2932.
-
Jeon H, Ono M, Kumagai C, Miki K, Morita A, et al. (1996) Pericytes from microvessel fragment produce type IV collagen and multiple laminin isoforms. Biosci Biotechnol Biochem 60: 856-861.
-
Katoh-Semba R, Kashiwamata S (1980) Interaction of bilirubin with brain capillaries and its toxicity. Biochim Biophys Acta 632: 290-297.
-
Fina L, Molgaard HV, Robertson D, Bradley NJ, Monaghan P, et al. (1990) Expression of the CD34 gene in vascular endothelial cells. Blood 75: 2417-2426.
-
Nag S, Manias JL, Stewart DJ (2009) Pathology and new players in the pathogenesis of brain edema. Acta Neuropathol 118: 197-217.
-
Garman RH (2011) Histology of the central nervous system. Toxicol Pathol 39: 22-35.
-
Schutta HS, Johnson L (1967) Bilirubin encephalopathy in the Gunn rat: a fine structure study of the cerebellar cortex. J Neuropathol Exp Neurol 26: 377-396.
-
Hatten ME, Heintz N (1995) Mechanisms of neural patterning and specification in the developing cerebellum. Annu Rev Neurosci 18: 385-408.
-
Bizzoca A, Virgintino D, Lorusso L, Buttiglione M, Yoshida L, et al. (2003) Transgenic mice expressing F3/contactin from the TAG-1 promoter exhibit developmentally regulated changes in the differentiation of cerebellar neurons. Development 130: 29-43.
-
Bauer-Moffett C, Altman J (1977) The effect of ethanol chronically administered to preweanling rats on cerebellar development: a morphological study. Brain Res 119: 249-268.
-
Herrup K, Sunter K (1987) Numerical matching during cerebellar development: quantitative analysis of granule cell death in staggerer mouse chimeras. J Neurosci 7: 829-836.
-
Robert MC, Furlan G, Rosso N, Gambaro SE, Apitsionak F, et al. (2013) Alterations in the cell cycle in the cerebellum of hyperbilirubinemic Gunn rat: a possible link with apoptosis? PLoS One 8: e79073.
-
Rosenberg GA (2012) Neurological diseases in relation to the blood-brain barrier. J Cereb Blood Flow Metab 32: 1139-1151.
-
Zlokovic BV (2011) Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders. Nat Rev Neurosci 12: 723-738.
-
Falcão AS, Silva RF, Vaz AR, Silva SL, Fernandes A, et al. (2013) Cross-talk between neurons and astrocytes in response to bilirubin: early beneficial effects. Neurochem Res 38: 644-659.
-
Falcão AS, Silva RF, Vaz AR, Gomes C, Fernandes A, et al. (2014) Cross-talk between neurons and astrocytes in response to bilirubin: adverse secondary impacts. Neurotox Res 26: 1-15.
-
Fernandes A, Falcão AS, Silva RF, Gordo AC, Gama MJ, et al. (2006) Inflammatory signalling pathways involved in astroglial activation by unconjugated bilirubin. J Neurochem 96: 1667-1679.
-
Palmela I, Cardoso FL, Bernas M, Correia L, Vaz AR, et al. (2011) Elevated levels of bilirubin and long-term exposure impair human brain microvascular endothelial cell integrity. Curr Neurovasc Res 8: 153-169.
-
de Vries HE, Blom-Roosemalen MC, van Oosten M, de Boer AG, van Berkel TJ, et al. (1996) The influence of cytokines on the integrity of the blood-brain barrier in vitro. J Neuroimmunol 64: 37-43.
-
Nakagawa S, Deli MA, Kawaguchi H, Shimizudani T, Shimono T, et al. (2009) A new blood-brain barrier model using primary rat brain endothelial cells, pericytes and astrocytes. Neurochem Int 54: 253-263.
-
Hellström M, Gerhardt H, Kalén M, Li X, Eriksson U, et al. (2001) Lack of pericytes leads to endothelial hyperplasia and abnormal vascular morphogenesis. J Cell Biol 153: 543-553.
-
Armulik A, Genové G, Mäe M, Nisancioglu MH, Wallgard E, et al. (2010) Pericytes regulate the blood-brain barrier. Nature 468: 557-561.
-
Winkler EA, Bell RD, Zlokovic BV (2010) Pericyte-specific expression of PDGF beta receptor in mouse models with normal and deficient PDGF beta receptor signaling. Mol Neurodegener 5: 32.
-
Hammes HP, Lin J, Renner O, Shani M, Lundqvist A, et al. (2002) Pericytes and the pathogenesis of diabetic retinopathy. Diabetes 51: 3107-3112.
-
Braun A, Xu H, Hu F, Kocherlakota P, Siegel D, et al. (2007) Paucity of pericytes in germinal matrix vasculature of premature infants. J Neurosci 27: 12012-12024.
-
Cardoso FL, Brites D, Brito MA (2010) Looking at the blood-brain barrier: molecular anatomy and possible investigation approaches. Brain Res Rev 64: 328-363.
-
Reyes R, Guo M, Swann K, Shetgeri SU, Sprague SM, et al. (2009) Role of tumor necrosis factor-alpha and matrix metalloproteinase-9 in blood-brain barrier disruption after peripheral thermal injury in rats. J Neurosurg 110: 1218-1226.
-
Haorah J, Schall K, Ramirez SH, Persidsky Y (2008) Activation of protein tyrosine kinases and matrix metalloproteinases causes blood-brain barrier injury: Novel mechanism for neurodegeneration associated with alcohol abuse. Glia 56: 78-88.
-
Muellner A, Benz M, Kloss CU, Mautes A, Burggraf D, et al. (2003) Microvascular basal lamina antigen loss after traumatic brain injury in the rat. J Neurotrauma 20: 745-754.
-
Hamann GF, Liebetrau M, Martens H, Burggraf D, Kloss CU (2002) Microvascular basal lamina injury after experimental focal cerebral ischemia and reperfusion in the rat. J Cereb Blood Flow Metab 22: 526-533.
-
Kalebic T, Garbisa S, Glaser B, Liotta LA (1983) Basement membrane collagen: degradation by migrating endothelial cells. Science 221: 281-283.
-
Watchko JF (2006) Kernicterus and the molecular mechanisms of bilirubin-induced CNS injury in newborns. Neuromolecular Med 8: 513-529.
-
Nag S, Venugopalan R, Stewart DJ (2007) Increased caveolin-1 expression precedes decreased expression of occludin and claudin-5 during blood-brain barrier breakdown. Acta Neuropathol 114: 459-469.
-
Kronstein R, Seebach J, Grossklaus S, Minten C, Engelhardt B, et al. (2012) Caveolin-1 opens endothelial cell junctions by targeting catenins. Cardiovasc Res 93: 130-140.
-
Zhong Y, Smart EJ, Weksler B, Couraud PO, Hennig B, et al. (2008) Caveolin-1 regulates human immunodeficiency virus-1 Tat-induced alterations of tight junction protein expression via modulation of the Ras signaling. J Neurosci 28: 7788-7796.
-
Nag S, Manias JL, Stewart DJ (2009) Expression of endothelial phosphorylated caveolin-1 is increased in brain injury. Neuropathol Appl Neurobiol 35: 417-426.
-
Beauchesne E, Desjardins P, Butterworth RF, Hazell AS (2010) Up-regulation of caveolin-1 and blood-brain barrier breakdown are attenuated by N-acetylcysteine in thiamine deficiency. Neurochem Int 57: 830-837.
-
Chang CF, Chen SF, Lee TS, Lee HF, Chen SF, et al. (2011) Caveolin-1 deletion reduces early brain injury after experimental intracerebral hemorrhage. Am J Pathol 178: 1749-1761.
-
Wang P, Liu Y, Shang X, Xue Y (2011) CRM197-induced blood-brain barrier permeability increase is mediated by upregulation of caveolin-1 protein. J Mol Neurosci 43: 485-492.
