

Journal of Rheumatic Diseases and Treatment
Suppression of Experimental Arthritis through AMP-Activated Protein Kinase Activation and Autophagy Modulation
Huimin Yan, Hui-fang Zhou, Ying Hu and Christine T.N. Pham*
Department of Medicine, Division of Rheumatology, Washington University School of Medicine, USA
*Corresponding author: Christine Pham, MD, Division of Rheumatology, Washington University School of Medicine, 660 S. Euclid Avenue, Campus Box 8045, Saint Louis, MO, 63110, USA, Tel: 314-362-9043, Fax: 314-454-1091, E-mail: cpham@dom.wustl.edu
J Rheum Dis Treat, JRDT-1-005, (Volume 1, Issue 1), Research Article; ISSN: 2469-5726
Received: January 31, 2015 | Accepted: February 25, 2015 | Published: February 28, 2015
Citation: Yan H, Zhou HF, Hu Y, Pham CTN (2015) Suppression of Experimental Arthritis through AMP-Activated Protein Kinase Activation and Autophagy Modulation J Rheum Dis Treat 1:005. 10.23937/2469-5726/1510005
Copyright: © 2015 Yan H, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Autophagy plays a central role in various disease processes. However, its contribution to inflammatory arthritides such as rheumatoid arthritis (RA) is unclear. We observed that autophagy is engaged in the K/BxN serum transfer model of RA but autophagic flux is severely impaired. Metformin is an anti-diabetic drug that has been shown to stimulate autophagy. Induction of autophagic flux, through metformin-mediated AMP-activated protein kinase (AMPK) activation and interruption of mammalian target of rapamycin (mTOR) signaling mitigated the inflammation in experimental arthritis. Further investigation into the effects of metformin suggest that the drug directly activates AMPK and dose-dependently suppressed the release of TNF-α, IL-6, and MCP-1 by macrophages while enhancing the release of IL-10 in vitro. In vivo, metformin treatment significantly suppressed clinical arthritis and inflammatory cytokine production. Mechanistic studies suggest that metformin exerts its anti-inflammatory effects by correcting the impaired autophagic flux observed in the K/BxN arthritis model and suppressing NF-κB-mediated signaling through selective degradation of IκB kinase (IKK). These findings establish a central role for autophagy in inflammatory arthritis and argue that autophagy modulators such as metformin may represent potential therapeutic agents for the treatment of RA.
Keywords
Autophagy, AMP-activated protein kinase (AMPK), Mammalian target of rapamycin (mTOR), Metformin, Inflammatory arthritis
Abbreviations
AMPK, 5: Adenosine Monophosphate-Activated Protein Kinase, Atg: Autophagy-Related Gene, ER: Endoplasmic Reticulum, IKK: I kappa B Kinase, IL: Interleukin, LAMP: Lysosomal-Associated Membrane Protein, LC3: Microtubule-Associated Light Chain 3, MCP-1: Monocyte Chemoattractant Protein 1, mTOR: Mammalian Target of Rapamycin, mTORC1: mTOR Complex 1, NF-?B: Nuclear Factor-Kappa B, RA: Rheumatoid Arthritis, RASF: RA Synovial Fibroblast, STAT: Signal Transducer and Activator of Transcription, SQSTM1: Sequestome1, TNF: Tumor Necrosis Factor, TRAF: TNF Receptor Associated Factor, ULK: Unc-51-like kinase.
Introduction
Rheumatoid arthritis (RA) is a complex inflammatory disease, characterized by an abundant cellular infiltration consisting of neutrophils, macrophages, and lymphocytes, leading to the release of multiple inflammatory cytokines and matrix-degrading enzymes that contribute to the progressive joint destruction [1]. These inflammatory cytokines, including TNF-α and IL-1β, have been reported to induce an endoplasmic reticulum (ER) stress response, leading to the accumulation of unfolded/misfolded, polyubiquitinated protein aggregates that can initiate and perpetuate inflammation [2,3]. Effective removal of these toxic factors can help relieve cell stress, reinstate ER homeostasis, and curtail the inflammation. Autophagy (also known as macroautophagy) is involved in a number of cellular homeostatic processes and plays a central role in the innate and adaptive immune response by recycling and removing harmful protein aggregates and damaged cell organelles [4,5]. The dysregulation of autophagic pathways has also been implicated in the pathogenesis of various disease processes, such as tumorigenesis and inflammation [6,7]. Autophagy has been implicated in RA; however, its exact contribution to disease manifestation is unclear. Metformin, a biguanide, is the first-line oral therapy for type 2 diabetes and the most widely used antidiabetic drug, alone or in combination with other antihyperglycemic agents [8]. Metformin acutely decreases hepatic glucose production through direct inhibition of the mitochondrial respiratory chain complex I [9]. More recently, much attention has been given to the effects of metformin in reducing cancer risk in patients with type 2 diabetes [10]. It has been proposed that metformin derived anti-cancer effects through the activation of AMP-activated protein kinase (AMPK) and negative regulation of mammalian target of rapamycin (mTOR) [11], both of which are implicated in the autophagy signaling network [12]. Since autophagy recently emerged as a central regulator in the induction and maintenance of inflammation [6], we sought to determine whether modulation of autophagy, through metformin-mediated AMPK activation, would mitigate the inflammation in experimental arthritis. We found that autophagy was indeed initiated in experimental arthritis; however, autophagic flux was severely impaired. Metformin-mediated inhibition of mTOR activity induced the progression of autophagic flux, which resulted in the suppression of NF-κB signaling and inflammatory cytokine production. These findings suggest a central role for autophagy in inflammatory arthritis and argue that autophagy modulators such as metformin may represent potential therapeutic agents for the treatment of RA.
Results
Metformin suppresses experimental arthritis
Experimental arthritis was induced with the passive transfer of serum from the KRN/I-Ag7 (K/BxN) mice [13]. The disease caused by arthrogenic autoantibodies in KRN serum shares many features with human RA [14]. WT C57BL/6 mice were injected i.p. with 175�l of KRN serum on day 0. Clinical arthritis was evident after 24 h and maximal disease developed around day 6-7. Metformin [150mg/kg] was injected i.p. every day, starting on day -1 and continued for the duration of the experiment. Metformin treatment significantly suppressed clinical disease compared with saline control (Figure 1A). In parallel with improved clinical scores, we observed on histological analysis a marked decrease in inflammatory cell influx, bone erosions, and cartilage degradation (Figure 1B,1C). Moreover, joint-associated inflammatory cytokines (TNF-α, IL-1β, IL-6, MCP-1) levels on day 9 were all significantly suppressed in the metformin treatment group (Figure 1D). Even when metformin treatment was delayed until day 2 post-KRN serum administration, when arthritis is established, we still observed significant suppression of disease activity (Figure 1E). Taken together these results suggest that metformin is effective at suppressing arthritis especially when started early or during the preclinical phase of the disease when there are elevations of disease-related biomarkers (i.e. anti-citrullinated peptide antibodies and/or rheumatoid factor) but no overt joint symptoms [15,16].
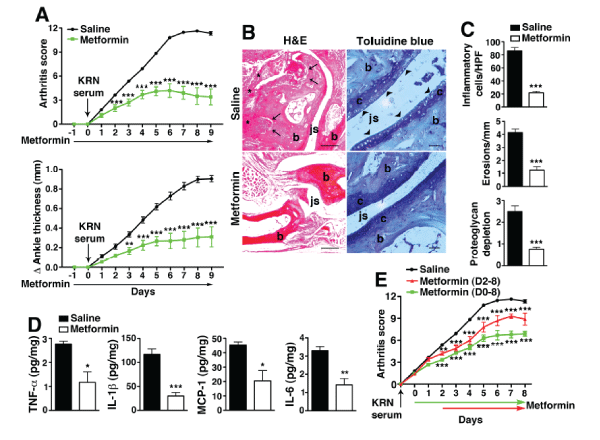
Figure 1: Metformin suppresses KRN arthritis.
(A) Cohorts of mice were injected i.p daily with saline or metformin (150 mg/kg of body weight) starting one day prior (day -1) to KRN serum transfer and
continued daily for the duration of the experiment. On day 0, they were injected i.p. with 175�l of KRN serum. Changes in arthritis score and ankle thickness were
assessed daily. (B) Representative micrographs of day 9 joint sections stained with H&E and toluidine blue. Scale bars=200�m (H&E), 50 �m (toluidine blue) (C)
Graphical representations of inflammatory cellular infiltrates, bone erosions and cartilage degradation. (D) Day 9 paws were homogenized and cleared lysates
were assayed for inflammatory cytokines. Levels are expressed as picograms (pg) per mg of total protein extracts. (E) Arthritis was induced with KRN serum
transfer on day 0 and metformin started on day 0 or day 2 after disease is established. Values are presented as mean � SEM, n=15 mice in the saline group and
5 mice for each metformin treatment group. *P< 0.05, **P< 0.01, ***P< 0.001.
View Figure 1
Metformin directly suppresses macrophage inflammatory activity in vitro
Previous studies suggest that metformin suppressed anti-collagen antibody-induced arthritis through down regulation of Th17 cell differentiation [17,18]. However, it has been shown that in the standard KRN serum transfer arthritis model, the downstream effector mechanisms involved in disease manifestation induced by arthrogenic autoantibodies do not require T or B lymphocytes nor are they dependent on IL-17 [19,20]. We thus reasoned that metformin might have a more direct effect on the innate immune response that initiated the inflammatory cascade in KRN serum transfer arthritis. A key study by Zhou et al. [21] suggests that metformin activates AMP-activated protein kinase (AMPK) [21], a major intracellular energy sensor and regulator of energy homeostasis [22]. AMPK has more recently been linked to the regulation of inflammatory signaling, especially macrophage inflammatory response [23,24]. Thus, we next evaluated the cytokine profile of inflammatory peritoneal macrophages ex vivo in response to metformin. Indeed, metformin dose-dependently suppressed the release of TNF-α, IL-6, and MCP-1 by macrophages while enhancing the release of IL-10 (Figure 2A). In parallel with the anti-inflammatory cytokine profile, we also observed a dose-dependent increase in the phosphorylation of AMPK (Figure 2B).
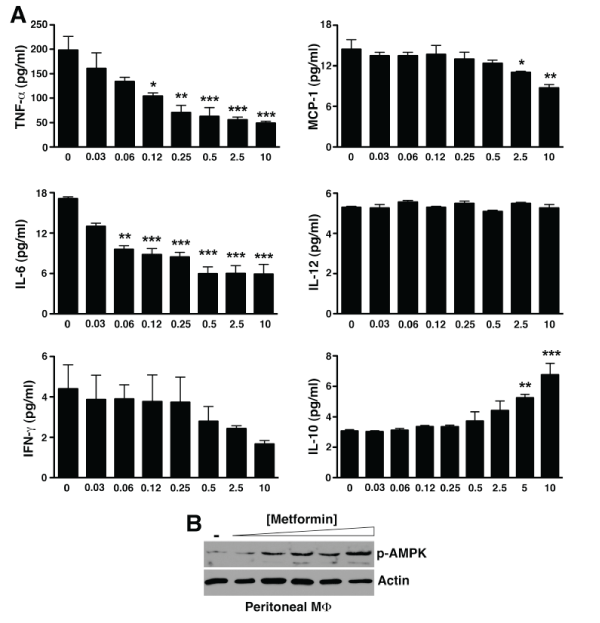
Figure 2: Metformin directly suppresses macrophage inflammatory activity in vitro.
(A) Day 5 thioglycollate-elicited peritoneal macrophages were cultured with the indicated metformin concentrations. At 48 h, supernatants were collected
and assayed for cytokines by cytometric bead arrays. Values represent mean � SEM of triplicate samples derived from 3 independent experiments. *P< 0.05,
**P< 0.01, ***P< 0.001 (B) Cultured macrophages were lysed, cleared, fractionated by SDS-PAGE, and probed for phospho-AMPK. Actin served as control for
protein loading.
View Figure 2
Metformin suppresses inflammatory macrophage phenotype in KRN arthritis
Macrophages are central to the pathogenesis of RA. Activated macrophages in RA synovium produce inflammatory cytokines including TNF-α, IL-1β, IL-6 [1], suggesting they are skewed toward the classically activated M1 phenotype via STAT1 activation [25]. Although alternatively activated [M2] macrophages are also found in RA synovium [26], the success of biologics such as anti-TNF agents suggests that the balance between pro- and anti-inflammatory cytokines is tipped toward M1 macrophages. Consistent with these findings we previously observed a predominance of CD206+ (mannose receptor, an M2 marker) macrophages in KRN arthritic paws [27]. Treatment with metformin, however, led to the suppression of TNF-α expression and STAT1 activation at the cellular level (Figure 3AB), suggesting that metformin exerts a counter-inflammatory effect on macrophage functional phenotype in vivo, thus corroborating the in vitro findings (Figure 2).
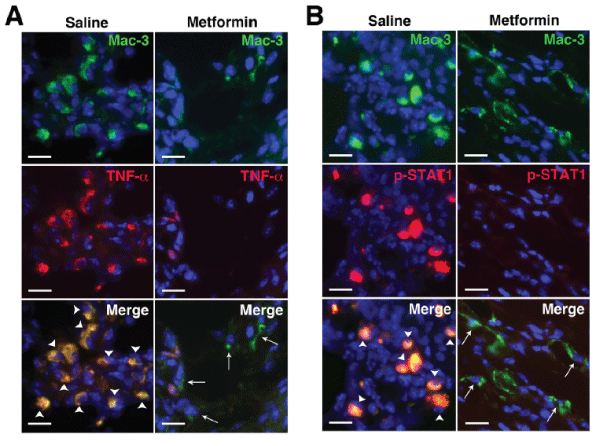
Figure 3: Metformin suppresses inflammatory macrophage phenotype in KRN arthritis.
Day 9 paws were stained for (A) macrophages (Mac-3, green) and TNF-a (red) and (B) Mac-3 (green) and phospho-STAT1 (red). In saline control animals there
was significant colocalization (yellow) of TNF-a and phospho-STAT1 with Mac-3+ cells (arrowheads) while metformin treatment suppressed both TNF-a and
STAT1 activation in macrophages (arrows). DAPI (blue) stained nuclei. Scale bar=25�m
View Figure 3
Metformin activates AMPK and suppresses mTOR activity in KRN arthritis
To determine whether metformin also modulates AMPK activity in vivo, paw lysates from treated animals were examined for AMPK phosphorylation by Western blot analysis. We observed increased AMPK phosphorylation in total paw lysates from metformin-treated animals that peaked on day 7 post KRN serum transfer (Figure 4A). The equivalent level of phospho-AMPK in total paw lysates on day 9 by Western blot analysis likely reflects the reduced number of inflammatory cells following metformin treatment (Figure 1C). Indeed, we confirmed that AMPK phosphorylation was still significantly enhanced on day 9 in synovial macrophages of metformin-treated compared with saline-treated animals (Figure 4B). AMPK is known to inhibit the activity of mTOR, a pathway that negatively regulates autophagy [12]. We observed a decrease in mTOR complex 1 (mTORC1) signaling, evidenced by decreased phosphorylation of mTOR and its downstream effector, ribosomal protein S6, in metformin-treated animals (Figure 4C,4D).
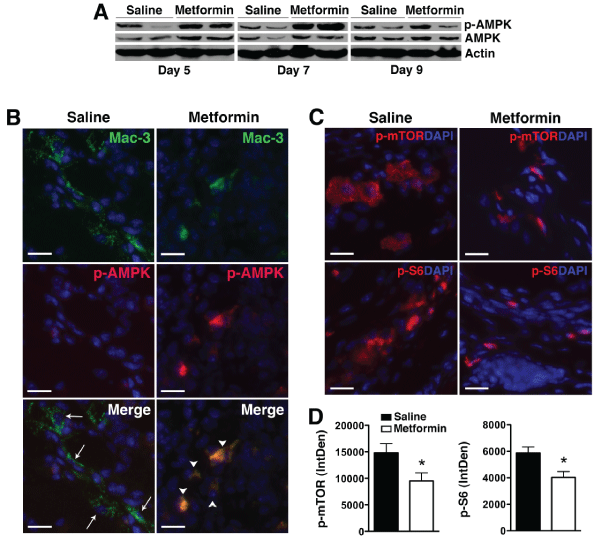
Figure 4: Metformin activates AMPK and suppresses mTORC1 activity in KRN arthritis.
(A) Protein lysates from day 5, 7, or 9 paws obtained from KRN arthritic mice were probed for p-AMPK and total AMPK. Actin served as control for protein loading.
(B) Day 9 paws were stained for Mac-3 (arrows, green) and p-AMPK (red). Colocalization (arrowheads) appeared orange/yellow. (C) Day 9 paws were stained for
phospho (p)-mTOR and p-S6 (red). DAPI (blue) stained nuclei. Scale bar=25�m (D) Intracellular level of p-mTOR and p-S6 was analyzed using ImageJ program
as detailed in the Materials and Methods section and presented as integrated optical density (IntDen). Values represent mean � SEM, n = 4-5 mice per treatment
group. *P<0.05 compared with saline.
View Figure 4
Metformin enhances autophagic flux
We next considered whether metformin-induced AMPK activation and mTOR inhibition enhanced autophagic activity. Although autophagy is implicated in RA [28,29], its specific contribution to disease manifestation is still unclear. We previously showed that autophagy was activated in KRN arthritis [27], as evidenced by upregulation of microtubule-associated light chain 3 [LC3] expression, conversion of LC3-I to LC3-II by lipidation (Figure 5A), and formation of autophagosomes, seen as punctate LC3+ immunofluorescent dots [30] and (Figure 5B). Although these findings indicate that autophagy was initiated during KRN arthritis, efficient progression of autophagy (autophagic flux) appeared to be impaired, evidenced by the accumulation of LC3-II and p62 (Figure 5A). p62, also known as SQSTM1/sequestome1, is an adaptor protein that participates in the delivery of ubiquinated proteins to autophagosomes. LC3-II and p62 are incorporated into autophagosomes and degraded during autophagic progression [30,31]. Thus, LC3-II and p62 levels inversely correlate with autophagic activity/flux. One explanation for the impaired autophagic flux in KRN arthritis could be the blockade of fusion between autophagosomes and lysosomes to form autolysosomes [32]. Indeed we found minimal co-localization between punctate LC3+ cells and LAMP-1 (a marker of lysosomes) (Figure 5B) in KRN arthritis. Metformin treatment, on the other hand, induced the formation of autolysosomes, as evidenced by colocalization of LC3 and LAMP-1 (Figure 5B), and the progression of autophagic flux, which was reflected in decreased LC3-II and p62 levels (Figure 5C). Initiation of the autophagic cascade also requires the mammalian unc-51-like kinase 1 (ULK1) complex [33]. AMPK (and mTORC1) are known to phosphorylate ULK1 at various sites but the effects of AMPK on ULK1 in the control of autophagy are still not well-established [33]. We found significantly lower level of phospho-ULK1Ser555 in day 9 metformin-treated paws, suggesting that dephosphorylation of ULK1 may represent an important event in the regulation of autophagic activity (Figure 5D).
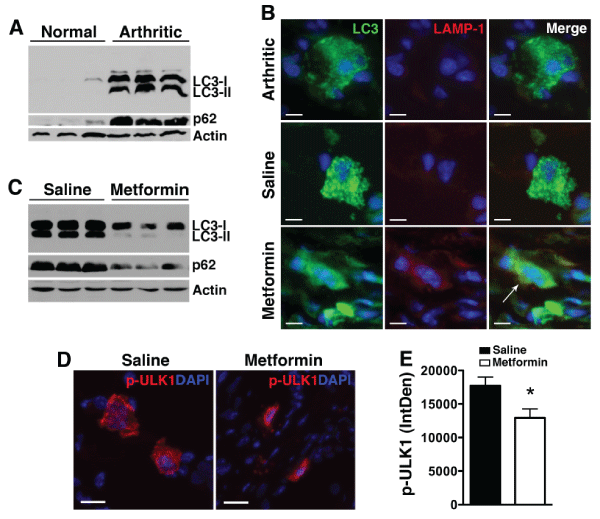
Figure 5: Metformin enhances autophagic flux.
(A) Paw lysates from normal and day 9 KRN serum-induced arthritic mice were probed for LC3-I, the lipidated form LC3-II, and p62. Actin served as control
for protein loading. (B) Paw sections from untreated arthritic mice or arthritic mice treated with saline or metformin were stained for LC3 (green) and the
lysosomal marker LAMP-1 (red). Note the punctate LC3 staining in untreated or saline control arthritic paws. Colocalization of LC3 and LAMP-1 (arrow) indicates
autolysosome. Scale bar=10�m (C) Treatment with metformin led to degradation of the lipidated LC3-II form and p62, indicating enhanced autophagic flux. (D)
Day 9 paws were stained for phospho (p)-ULK1Ser555 (red). DAPI (blue) stained nuclei. Scale bar=25�m (E) Intracellular level of p-ULK1was analyzed using
ImageJ program as detailed in the Materials and Methods section and presented as integrated optical density (IntDen). Values represent mean � SEM, n=4-5
mice per treatment group. *P< 0.05 compared with saline.
View Figure 5
Metformin promotes degradation of NF-κB protein Iκ kinase (IKK) through autophagy
There is considerable new evidence pointing to a role of autophagy in the clearance of cytotoxic protein aggregates that accumulate in disease states and alterations in autophagy-dependent events result in the excessive production of inflammatory cytokines [6]. We reasoned that metformin-induced AMPK activation enhanced autophagic progression and the increased flux mediated protein degradation through the (auto) lysosomal pathway, thus suppressing the inflammatory responses. NF-κB plays a central role in the pathogenesis of RA [34]. The NF-κB family comprises five members: p105 (constitutively processed to p50), p100 (processed to p52 under regulated conditions), p65 (also known as RelA), RelB, and c-Rel [35]. These members form homo- and heterodimers that, in the resting cell, are normally held inactive in the cytoplasm by the association with inhibitors, the IκB proteins. Upstream signaling and activation of the NF-κB pathway converge on the IκB kinase (IKK) complex that phosphorylates IκB proteins and targets them for degradation, releasing the NF-κB Rel subunits for nuclear translocation and transactivation of a multitude of responsive genes, including several inflammatory cytokines. Recent studies suggest a tight regulation between NF-κB pathway and autophagy [36,37]. Indeed Qing et al. [38] have shown that IKK proteins are substrates for the autophagy pathway [38]. Thus, autophagic degradation of IKK would potentially limit the role of NF-κB in inflammation. We examined various NF-κB proteins in day 9 paw lysates and observed specific decrease in IKK(α) level with relative preservation of IκB and p65 protein expression (Figure 6A) in metformin-treated animals. IKK(α) degradation suppressed NF-κB signaling, as evidenced by depressed p65 phosphorylation/activation following metformin treatment (Figure 6A). We further investigated how metformin induced IKK degradation. During the cellular stress that accompanies inflammatory arthritis, large quantities of newly synthesized proteins are polyubiquinated [39] and recognized by p62/ SQSTM1 (Figure 6B). In fact, p62-polyubiquinated protein aggregates have recently been described in RA synovial cells [29]. We found that IKKα was indeed ubiquinated (Figure 6C) and, following metformin treatment, co-localized to the LAMP- 1+ compartment (Figure 6D), suggesting delivery to the lysosomal pathway (perhaps through p62 association) for degradation. On the other hand, impaired autophagic flux [in saline controls] led to the accumulation of IKK (Figure 6D). Taken together these results suggest that metformin corrected the impaired autophagic flux in KRN arthritis and suppressed NF-κB-induced inflammation, at least partly through selective IKK degradation.
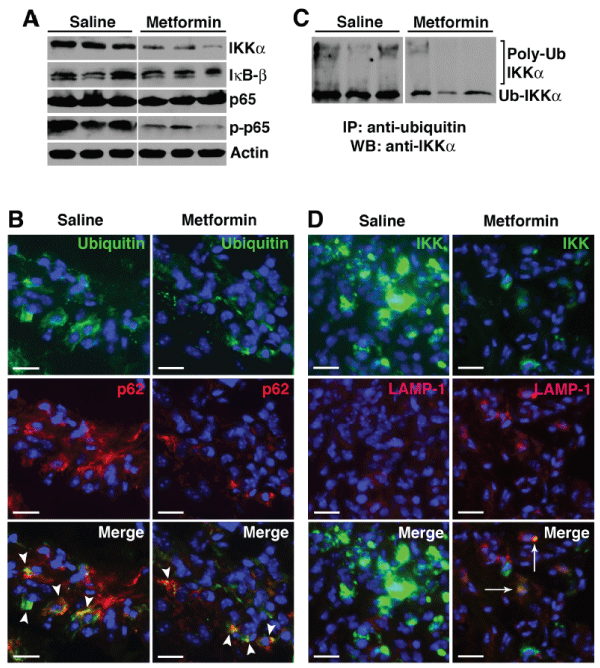
Figure 6: Metformin promotes selective degradation of NF-?B protein I? kinase (IKK) through autophagy.
(A) Day 9 paw lysates from saline controls and metformin-treated animals were probed for NF-?B proteins (IKKa, I?B-�, p65, and phospho-p65). Actin served as
protein loading control. (B) Day 9 paw sections were probed for ubiquitin (green) and p62 (red). Colocalization (arrowheads, yellow) suggests ubiquinated-p62
aggregates. (C) Equivalent amounts of protein were immunoprecipitated with anti-ubiquitin antibody and probed with anti- IKKa antibody. High molecular weight
protein complexes likely represent poly-ubiquinated IKKa. (D) Paw sections were also probed for IKKa and LAMP-1. Accumulation of IKKa was evident in
the saline controls while IKKa level was significantly lower in metformin-treated animals. Colocalization of IKKa and LAMP-1 (arrows) in metformin treatment
suggests IKKa targeted to the lysosomes.
View Figure 6
Discussion
We show herein that metformin stimulates macrophage AMPK activity in KRN arthritis and this stimulation inhibits the activity of mTORC1, enhancing autophagic flux and decreasing inflammatory cytokine production through STAT1 suppression and selective degradation of IKK through the (auto) lysosomal pathway. Metformin has been attracting increased attention of late, mainly as an anti-cancer agent, alone or in combination with cytotoxic therapy [40]. These studies indicate that the direct effects of metformin on cancer cells are partially dependent on the AMPK-mTOR-signaling axis [40]. On the other hand the role of metformin in autophagy in RA has not been extensively explored. Limited studies previously suggest that phenformin, a biguanide with similar activity to metformin, improved clinical disease and decreased ESR in patients with RA [41]. Phenformin, however, was mostly withdrawn from the world markets due to safety issues [42]. More recent reports examining the effect of metformin in anti-collagen antibody-mediated arthritis suggests that the drug was effective in knocking down inflammation, although the authors proposed a mechanism dependent on down modulation of Th-17 cells [17,18]. The effector mechanisms in autoantibody-mediated arthritis induced by passive transfer of KRN serum, however, has been shown to be independent of T and B cells and IL-17 [19,20]. Metformin has been in clinical use for over 50 years and is the first line oral therapy for type 2 diabetes due partly to an impressive safety record [43]. Although extensively used, the molecular mechanism of action of metformin was not established until a key study was performed by Zhou et al. demonstrating that metformin activates AMPK, a major cellular energy sensor and regulator of homeostasis [21]. Moreover, AMPK activity in macrophages has been shown to suppress inflammatory responses and promote macrophage polarization toward an anti-inflammatory phenotype [23]. Thus AMPK activation is predominantly anti-inflammatory, likely occurring through mTOR inhibition and autophagy modulation [6]. We confirmed that metformin treatment directly activated AMPK in macrophages and inhibited mTORC1 activity in KRN arthritis. These signaling events led to enhanced autophagic flux.
Autophagy studies in RA have been focusing mainly on osteoclastic activity using in vitro assays. Deletion of autophagy gene Atg7 in TNF-α transgenic mice reduced the number of osteoclasts and mitigated joint damage [44]. Autophagy has also been studied in the context of RA synovial fibroblast survival. Kato et al. [29] examined RA synovial fibroblasts (RASFs) ex vivo and found that RASFs are hypersensitive to autophagic cell death under conditions of severe ER stress [29]. However, the role of autophagy in the modulation of inflammation in RA and experimental arthritis has not been extensively examined. We observed that, although autophagy was initiated in KRN arthritis, autophagic flux appeared severely impaired, as evidenced by elevated levels of lipidated LC3-II and p62 and accumulation of ubiquinated proteins. Decline in autophagic flux impaired cellular clearance and stimulated NF-κB signaling, generating chronic inflammation [36]. On the other hand, IKK proteins are known substrates of autophagy [38] and degradation of IKK would limit the role of NF-κB in inflammation. In summary, impaired autophagy in KRN arthritis perpetuated the chronic cycle of inflammation while IKK degradation following metformin treatment suppressed NF-κB-mediated inflammation.
The accumulation of p62, a cargo receptor for degradation of ubiquinated proteins through the autophagy pathway, stimulates the activation of NF-κB in response to several stimuli including TNF-α and IL-1β [45]. In addition to its role in trafficking ubiquinated proteins for autophagic degradation, p62 is also involved in various signaling events. p62 is required for the signal transduction pathways activated by TNF receptor associated factor 6 (TRAF-6) and p62-TRAF-6-mediated regulation of NF-κB [46]. How p62-TRAF-6 complex regulates NF-κB is not clear but may involve non-degradative ubiquination of IKKγ as p62 silencing abrogates TRAF-6-dependent ubiquination of IKKγ and severely impairs NF-κB activation [47]. Lastly, p62-TRAF-6 complex is also a critical regulator of mTOR activity and autophagy [48].
Studies examining the contribution of autophagy in RA and autophagy modulation through pharmacotherapeutics for the treatment of RA are limited. A recent report suggests that mTOR inhibition with the rapalog everolimus suppressed synovitis and protected against bone/cartilage damage in experimental arthritis [49]. These results corroborated a previous study showing superior clinical benefits with a combination of everolimus and methotrexate versus methotrexate alone in the treatment of RA [50]. These studies and our results herein suggest that modulation of autophagy [through direct mTOR inhibition or indirectly through AMPK activation] merits exploration as alternative treatment strategy in RA and potentially other inflammatory arthritides, especially since long-term use of rapamycin or rapalogs carries significant risk of immunosuppression unless tissue [joint]-specific delivery can be achieved.
In summary, we have shown that metformin activates AMPK and inhibits mTOR, correcting the impaired autophagic flux in KRN arthritis and suppressing NF-κB signaling. Because of the central role of NF-κB in immune responses, long-term general inhibition of this pathway may severely compromise the host�s ability to fight infection. The discovery that NF-κB-mediated inflammatory responses in the joints can be indirectly modulated by metformin through autophagy offers an alternative way to bypass general inhibition of NF-κB signaling.
Materials and Methods
K/BxN arthritis model and treatment
Six to eight week-old wild type C57BL/6 mice (The Jackson Laboratory, Bar Harbor, ME, USA) were injected i.p. with 175-200�l of KRN serum on day 0 and 1 to induce arthritis. Clinical disease was scored macroscopically on a scale ranging from 0�3 as previously described [51].
Paw thickness was measured daily by dial calipers and an average change in ankle thickness from the two hind paw measurements was determined for each mouse. Mice were randomly assigned to saline control or metformin (Cat # 2864, TOCRIS Bioscience) treatment administered i.p. daily at 150mg/kg body weight starting on day -1 and day 0 or 200 mg/kg starting on day 2 following KRN serum transfer.
Immunofluorescence
Frozen-sections of paws were fixed in 4% PFA, blocked in 8% BSA/PBS and incubated with the following primary antibodies: rabbit anti-mouse phospho-STAT1 (1:200, Cat # 9171, Cell Signaling Technology), IKK-alpha (1:200, Cat # 2682, Cell Signaling Technology), phospho-p65 (1:200, phosphorylation site S536, Cat # 3033, Cell Signaling Technology), phospho-mTOR (1:200; Cat# 5536, Cell Signaling Technology), phospho-S6 (1:200; Cat #4858, Cell Signaling Technology), LC3 (1:200, Cat # L7543, Sigma-Aldrich), phospho-ULK1 (1:200, Cat # 5869, Cell Signaling Technology), rat anti-mouse LAMP-1 (1:100, Cat # SC19992, Santa Cruz), mouse anti-mouse p62 (IgM, 1:500, Cat # MABC32, clone 11C9.2, Millipore), biotin-conjugated anti-mouse Mac-3 monoclonal antibody (1:200, Cat # CL8943AP, Cedarlane Laboratories), mouse anti-mouse ubiquitin (IgG, 1:500, Cat # 64301, clone P4D1, Biolegend) for 1 h at room temperature followed by the appropriate secondary antibodies: rhodamine red-conjugated secondary antibody (1:100, Jackson ImmunoResearch Laboratories), FITC conjugated-secondary antibody (1:100, Jackson ImmunoResearch Laboratories) or FITC conjugated-streptavidin (1:100, Cat: 7100-02, Southern Biotechnology Associates). All images were visualized on a Nikon Eclipse microscope and acquired with QCapture software.
Quantitative analysis of immunofluorescence
All images were visualized on a Nikon Eclipse fluorescence microscope and acquired with QCapture software using the same exposure time. The single-color images were loaded into Image J software (http://rsb.info.nih.gov/ij/) for analysis. Threshold brightness of the image was adjusted to facilitate selection of regions of interest (ROIs) and free hand tool used to eliminate nonspecific staining. All images were set with the same hue, saturation, and brightness for further image analysis/measurement. The data represent average obtained from 4-5 mice per treatment group, 10-15 fields per paw section and 5-6 sections per paw and presented as integrated optical density (IntDen).
Histological analysis
Paws were harvested on day 9 after serum transfer, fixed in 10% buffered formalin for 48 h before decalcification in EDTA solution and processing for paraffin embedding. The sections (5�m) were stained with Hematoxylin and Eosin (H&E) or toluidine blue. Inflammatory cells infiltrating the synovial lining and the joint cavity were enumerated in 8-10 random fields per section using H&E images acquired at 400x magnification. Proteoglycan depletion was scored on toluidine blue-stained sections on a scale from 0�3, ranging from fully stained cartilage (score=0) to fully destained cartilage (score = 3) as previously described [51]. The number of bone erosions was enumerated per mm of bone surface using ImageJ program (http://rsb.info.nih.gov/ij/). Scoring was performed by an observer blinded to the treatment.
Culture of peritoneal macrophages
Peritonitis was induced by i.p. injection of sterile thioglycollate (4 % w/v in 1mL of sterile saline). The peritoneal macrophages were isolated from the peritoneal lavage on day 5 as previously described [27]. The recovered macrophages were washed with sterile PBS, counted and seeded in triplicates at 2�105 cells per well on 96-well plate. The cells were allowed to adhere for 1 h at 37�C and non-adherent cells were removed with gentle washing. Serial doses of metformin (0.03, 0.06, 0.12, 0.25, 0.5, 2.5 and 10mM) were added and the cells were cultured at 37�C in 5% CO2 atmosphere. The supernatant was collected at 48 h for cytokine analysis and the cell lysates were prepared for Western blotting.
Cytokine analysis
Paws were homogenized in 1 ml of PBS with proteinase inhibitor cocktail and cleared by centrifugation. Cytokine concentrations were measured in equivalent volumes of paw lysates or culture supernatant using an ELISA kit for IL-1β (R&D Systems Inc., Minneapolis, Minnesota, USA) and cytometric bead Array (CBA, Mouse Inflammatory Cytokines kit, BD Bioscience) for TNF-α, IL-6, IL-10, IL-12p70, MCP-1 and INF-γ according to the manufacturer�s recommendations.
Western blot analysis
Snap-frozen paws or macrophages were processed in 1% NP-40 lysis buffer containing proteinase inhibitor cocktail (Sigma-Aldrich) and 50mM sodium fluoride (Sigma-Aldrich) and protein lysates cleared by centrifugation. Equivalent amounts of protein were resolved on a 10% SDS-PAGE gel and transferred to PVDF membrane (Millipore). The membrane was probed using the following primary antibodies: rabbit anti-mouse LC3 (1:2,000, Cat # L7543, Sigma-Aldrich), phospho-AMPKα (1:1,000, Cat # 2535, Cell Signaling Technology), phospho-mTOR (1:1,000, Cat # 5536, Cell Signaling Technology), IKK-α (1:1,000, Cat # 2682, Cell Signaling Technology), IκB-β (1:200, Cat # SC-945, Santa Cruz), total p65 (1:2,000, Cat # 8242, Cell Signaling Technology), phospho-p65 (1:2,000, Cat # 3033, Cell Signaling Technology) and mouse anti-mouse p62 (1:2,000, Cat # MABC32, clone 11C9.2, Millipore) followed by the appropriate HRP-conjugated secondary antibodies. Goat anti-mouse actin (1:3,000, Cat # SC-1615, Santa Cruz) served as control for quantity and quality of protein.
Immunoprecipitation of ubiquinated proteins
Paw protein lysates were prepared as described above. Equivalent amounts of precleared protein lysate (200�g and 20�l of protein G Sepharose beads (Sigma-Aldrich; Cat# P3296; in 20% ethanol) were incubated with monoclonal anti-ubiquitin antibody (IgG, 1:200, Cat # 64301, clone P4D1, Biolegend) overnight at 4�C under rotary agitation. The beads were washed three times in NP-40 buffer, boiled at 95-100�C for 5 min in SDS buffer, and fractionated on a SDS-PAGE gel. The proteins were transferred to PVDF membrane and probed with anti-IKK-α antibody (1:1,000, Cat # 2682, Cell Signaling Technology).
Statistical analysis
Comparisons between two groups and multiple groups (≥3) were performed by t-test and 2-way ANOVA, respectively, followed by Bonferroni post hoc test to compare all groups of data. Numerical data were expressed as mean � SEM. P values < 0.05 were considered significant.
Ethical Statement for Animal Research
All animal experiments were performed in compliance with federal laws and in strict accordance with the guidelines established by the Division of Comparative Medicine at Washington University. The animal protocol is subjected to annual review and approval by The Animal Studies Committee of Washington University.
Acknowledgments
This work was supported partially by NIH grants HL073646 and AI051436 and a bridge fund from the Department of Medicine.
Declaration
Huimin Yan and Hui-fang Zhou contributed equally to this work.
References
-
McInnes IB, Schett G (2007) Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol 7: 429-442.
-
Zhang K, Shen X, Wu J, Sakaki K, Saunders T, et al. (2006) Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell 124: 587-599.
-
Zhang K, Kaufman RJ (2008) From endoplasmic-reticulum stress to the inflammatory response. Nature 454: 455-462.
-
Schmid D, M�nz C (2007) Innate and adaptive immunity through autophagy. Immunity 27: 11-21.
-
M�nz C (2009) Enhancing immunity through autophagy. Annu Rev Immunol 27: 423-449.
-
Jones SA, Mills KH, Harris J (2013) Autophagy and inflammatory diseases. Immunol Cell Biol 91: 250-258.
-
Kenific CM, Debnath J2 (2015) Cellular and metabolic functions for autophagy in cancer cells. Trends Cell Biol 25: 37-45.
-
Ferrannini E (2014) The target of metformin in type 2 diabetes. N Engl J Med 371: 1547-1548.
-
Viollet B, Guigas B, Sanz Garcia N, Leclerc J, Foretz M, et al. (2012) Cellular and molecular mechanisms of metformin: an overview. Clin Sci (Lond) 122: 253-270.
-
Snima KS, Pillai P, Cherian AM, Nair SV, Lakshmanan VK1 (2014) Anti-diabetic drug metformin: challenges and perspectives for cancer therapy. Curr Cancer Drug Targets 14: 727-736.
-
Miranda VC, Barroso-Sousa R1, Glasberg J1, Riechelmann RP2 (2014) Exploring the role of metformin in anticancer treatments: a systematic review. Drugs Today (Barc) 50: 623-640.
-
Alers S, L�ffler AS, Wesselborg S, Stork B (2012) Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol 32: 2-11.
-
Kouskoff V, Korganow AS, Duchatelle V, Degott C, Benoist C, et al. (1996) Organ- specific disease provoked by systemic autoimmunity. Cell 87: 811-22.
-
Ditzel HJ (2004) The K/BxN mouse: a model of human inflammatory arthritis. Trends Mol Med 10: 40-45.
-
Nielen MM, van Schaardenburg D, Reesink HW, van de Stadt RJ, van der Horst-Bruinsma IE, et al. (2004) Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis Rheum 50: 380-386.
-
Majka DS, Deane KD, Parrish LA, Lazar AA, Bar�n AE, et al. (2008) Duration of preclinical rheumatoid arthritis-related autoantibody positivity increases in subjects with older age at time of disease diagnosis. Ann Rheum Dis 67: 801-807.
-
Kang KY, Kim YK, Yi H, Kim J, Jung HR, et al. (2013) Metformin downregulates Th17 cells differentiation and attenuates murine autoimmune arthritis. Int Immunopharmacol 16: 85-92.
-
Son HJ, Lee J, Lee SY, Kim EK, Park MJ, et al. (2014) Metformin attenuates experimental autoimmune arthritis through reciprocal regulation of Th17/Treg balance and osteoclastogenesis. Mediators Inflamm 2014: 973986.
-
Korganow AS, Ji H, Mangialaio S, Duchatelle V, Pelanda R, et al. (1999) From systemic T cell self-reactivity to organ-specific autoimmune disease via immunoglobulins. Immunity 10: 451-461.
-
Jacobs JP, Wu HJ, Benoist C, Mathis D (2009) IL-17-producing T cells can augment autoantibody-induced arthritis. Proc Natl Acad Sci U S A 106: 21789-21794.
-
Zhou G, Myers R, Li Y, Chen Y, Shen X, et al. (2001) Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 108: 1167-1174.
-
Hardie DG, Ross FA, Hawley SA (2012) AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 13: 251-262.
-
Sag D, Carling D, Stout RD, Suttles J (2008) Adenosine 5'-monophosphate-activated protein kinase promotes macrophage polarization to an anti-inflammatory functional phenotype. J Immunol 181: 8633-8641.
-
Yang Z, Kahn BB, Shi H, Xue BZ (2010) Macrophage alpha1 AMP-activated protein kinase (alpha1AMPK) antagonizes fatty acid-induced inflammation through SIRT1. J Biol Chem 285: 19051-19059.
-
Sica A, Mantovani A (2012) Macrophage plasticity and polarization: in vivo veritas. J Clin Invest 122: 787-795.
-
Kinne RW, Stuhlm�ller B, Burmester GR (2007) Cells of the synovium in rheumatoid arthritis. Macrophages. Arthritis Res Ther 9: 224.
-
Zhou HF, Yan H, Hu Y, Springer LE, Yang X, et al. (2014) Fumagillin prodrug nanotherapy suppresses macrophage inflammatory response via endothelial nitric oxide. ACS Nano 8: 7305-7317.
-
Shin YJ, Han SH, Kim DS, Lee GH, Yoo WH, et al. (2010) Autophagy induction and CHOP under-expression promotes survival of fibroblasts from rheumatoid arthritis patients under endoplasmic reticulum stress. Arthritis Res Ther 12: R19.
-
Kato M, Ospelt C, Gay RE, Gay S, Klein K (2014) Dual role of autophagy in stress-induced cell death in rheumatoid arthritis synovial fibroblasts. Arthritis Rheumatol 66: 40-48.
-
Mizushima N (2004) Methods for monitoring autophagy. Int J Biochem Cell Biol 36: 2491-2502.
-
Bj�rk�y G, Lamark T, Brech A, Outzen H, Perander M, et al. (2005) p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 171: 603-614.
-
Levine B, Kroemer G (2008) Autophagy in the pathogenesis of disease. Cell 132: 27-42.
-
Wong PM, Puente C, Ganley IG, Jiang X (2013) The ULK1 complex: sensing nutrient signals for autophagy activation. Autophagy 9: 124-137.
-
Simmonds RE, Foxwell BM (2008) Signalling, inflammation and arthritis: NF-kappaB and its relevance to arthritis and inflammation. Rheumatology (Oxford) 47: 584-590.
-
Pasparakis M (2009) Regulation of tissue homeostasis by NF-kappaB signalling: implications for inflammatory diseases. Nat Rev Immunol 9: 778-788.
-
Djavaheri-Mergny M, Codogno P (2007) Autophagy joins the game to regulate NF-kappaB signaling pathways. Cell Res 17: 576-577.
-
Trocoli A, Djavaheri-Mergny M (2011) The complex interplay between autophagy and NF-κB signaling pathways in cancer cells. Am J Cancer Res 1: 629-649.
-
Qing G, Yan P, Xiao G (2006) Hsp90 inhibition results in autophagy-mediated proteasome-independent degradation of IkappaB kinase (IKK). Cell Res 16: 895-901.
-
Yagishita N, Yamasaki S, Nishioka K, Nakajima T (2008) Synoviolin, protein folding and the maintenance of joint homeostasis. Nat Clin Pract Rheumatol 4: 91-97.
-
Pernicova I, Korbonits M1 (2014) Metformin--mode of action and clinical implications for diabetes and cancer. Nat Rev Endocrinol 10: 143-156.
-
Fearnley GR, Chakrabarti R (1966) Fibrinolytic treatment of rheumatoid arthritis with phenformin plus ethyloestrenol. Lancet 2: 757-761.
-
Luft D, Schm�lling RM, Eggstein M (1978) Lactic acidosis in biguanide-treated diabetics: a review of 330 cases. Diabetologia 14: 75-87.
-
Rena G, Pearson ER, Sakamoto K (2013) Molecular mechanism of action of metformin: old or new insights? Diabetologia 56: 1898-1906.
-
Lin NY, Beyer C, Giessl A, Kireva T, Scholtysek C, et al. (2013) Autophagy regulates TNFα-mediated joint destruction in experimental arthritis. Ann Rheum Dis 72: 761-768.
-
Moscat J, Diaz-Meco MT (2009) p62 at the crossroads of autophagy, apoptosis, and cancer. Cell 137: 1001-1004.
-
Moscat J, Diaz-Meco MT, Wooten MW (2007) Signal integration and diversification through the p62 scaffold protein. Trends Biochem Sci 32: 95-100.
-
Zotti T, Scudiero I1, Settembre P2, Ferravante A1, Mazzone P2, et al. (2014) TRAF6-mediated ubiquitination of NEMO requires p62/sequestosome-1. Mol Immunol 58: 27-31.
-
Linares JF, Duran A, Yajima T, Pasparakis M, Moscat J, et al. (2013) K63 polyubiquitination and activation of mTOR by the p62-TRAF6 complex in nutrient-activated cells. Mol Cell 51: 283-296.
-
Cejka D, Hayer S, Niederreiter B, Sieghart W, Fuereder T, et al. (2010) Mammalian target of rapamycin signaling is crucial for joint destruction in experimental arthritis and is activated in osteoclasts from patients with rheumatoid arthritis. Arthritis Rheum 62: 2294-2302.
-
Bruyn GA, Tate G, Caeiro F, Maldonado-Cocco J, Westhovens R, et al. (2008) Everolimus in patients with rheumatoid arthritis receiving concomitant methotrexate: a 3-month, double-blind, randomised, placebo-controlled, parallel-group, proof-of-concept study. Ann Rheum Dis 67: 1090-1095.
-
Zhou HF, Chan HW, Wickline SA, Lanza GM, Pham CT (2009) Alphavbeta3-targeted nanotherapy suppresses inflammatory arthritis in mice. FASEB J 23: 2978-2985.
