

Journal of Rheumatic Diseases and Treatment
Vogt-Koyanagi-Harada-Syndrome Concurrent with Morbus Darier
Claudia Eve Thieme1, Sibylle Winterhalter2, Bianca Apitzsch1 and Nicole Stuebiger1*
1Department of Ophthalmology, Charité, Campus Benjamin Franklin, Berlin, Germany
2Department of Ophthalmology, Charité, Campus Virchow, Berlin, Germany
*Corresponding author: Nicole Stuebiger, MD, Department of Ophthalmology, Charité, Campus, Benjamin Franklin, Hindenburgdamm 30, 12203 Berlin, Germany, Tel: +49 30 8445 2331, Fax: +49 30 8445 4450, E-mail: nicole.stuebiger@charite.de
J Rheum Dis Treat, JRDT-2-036, (Volume 2, Issue 2), Case Report; ISSN: 2469-5726
Received: April 15, 2016 | Accepted: May 31, 2016 | Published: June 02, 2016
Citation: Thieme CE, Winterhalter S, Apitzsch B, Stuebiger N (2016) Vogt-Koyanagi-Harada-Syndrome Concurrent with Morbus Darier. J Rheum Dis Treat 2:036. 10.23937/2469-5726/1510036
Copyright: © 2016 Thieme CE, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Keywords
Vogt-Koyanagi-Harada-Syndrome, Panuveitis, Morbus darier
Introduction
Vogt-Koyanagi-Harada-syndrome (VKH) is a multisystemic, T-cell mediated autoimmune disorder which is characterized by bilateral granulomatous panuveitis with extraocular manifestations in the central nervous and auditory system as well as in the integumentary field. Neurological and auditory manifestations include aseptic meningitis, meningism, tinnitus and cerebrospinal fluid pleocytosis. Integumentary findings can include alopecia, poliosis and vitiligo. The etiology of this disease is thought to be an immune reaction against melanocytes [1].
Although Vogt-Koyanagi-Harada-syndrome has not yet been described in combination with Morbus Darier, it is known that it can be accompanied by other immunological mediated inflammatory diseases such as polychondritis, Basedow’s disease and systemic sclerosis.
Herein we present - to our knowledge - the first description worldwide of a patient with incomplete Vogt-Koyanagi-Harada-syndrome (according to the New Diagnostic Criteria VKH 2001) previously diagnosed with Morbus Darier. According to the New Diagnostic Criteria, incomplete VKH is described as absent history of ocular trauma, no evidence for other ocular or systemic diseases and the presence of bilateral ocular disease (as early manifestation: diffuse chorioiditis with either focal subretinal fluid or bullous serous subretinal detachment or as late manifestation: ocular depigmentation, clumping in the retinal pigment epithelium, recurrent or chronic anterior uveitis) accompanied by either neurological or integumental findings. Morbus Darier is a rare genetic disease in which pustulous lesions occur at a juvenile age. Its occurrence can be sporadic, but it is also known to be associated with an autosomal-dominant mutation of the ATP2A2-gene on chromosome 12 (SERCA1-3-gene on chromosome 12q23-q24.1). Vogt-Koyanagi-Harada-syndrome is associated with HLA DRB1*0405 at the MHC-II locus on chromosome 6 [2]. Clinically, Morbus Darier shows scabby papules which are histologically characterized by acantholysis and precocious keratinization [3]. Patients suffer from itchiness after exposure to UV-light, sweating and in humidity. Morbus Darier is often accompanied by mental disorders such as epilepsy, mental retardation, depression and schizophrenia.
Case Report
A 52-year-old female patient presented with blurry vision and epiphora in both eyes of 3 weeks duration. There was no history of ocular trauma. She had low grade mental retardation and had been diagnosed with Morbus Darier by immunohistopathological testing 21 years ago. The histological findings, taken from a skin biopsy of the left submammary area and of the left Musculus pectoralis minor area, were typical for Morbus Darier and reported an acantholytical widened epithelium with basal and suprabasal disruptions. Moreover, corps ronds and grains were present in the epidermis.
Besides clinical examination, fluorescein angiography (FA), indocyanine green angiography (ICGA) were performed and optical coherence tomography (OCT) was done by using the spectral domain OCT (Heidelberg Engineering).
Serological testing included Toxoplasma gondii, Borrelia burgdorferi, ANA, c-ANCA and p-ANCA. HLA typing was processed from serology sample by sequence-specific oligonucleotides polymerase-chain reaction (SSO-PCR) with low resolution.
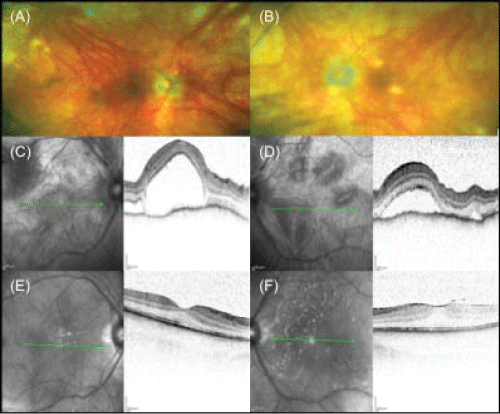
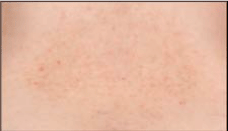
A bilateral panuveitis with optic disc hyperemia and edema was present and optical coherence tomography revealed a central serous retinal detachment with retinal folds in both eyes (Figure 1C and Figure 1D). In addition the patient disclosed anterior chamber (AC) inflammation with bilateral non-granulomatous retrocorneal precipitates and AC cells. FA showed dye pooling corresponding to areas in retinal detachments and punctual areas of hypofluorescence ("dark spots") were found in ICGA. Serological tests for Toxoplasma gondii, Borrelia burgdorferi, ANA, c-ANCA and p-ANCA were negative. The C-reactive protein was not elevated. Investigation of the HLA-DRB1 locus revealed no evidence of mutation. The patient was diagnosed as incomplete Vogt-Koyanagi-Harada-syndrome, according to the New Diagnostic Criteria [4,5]. Treatment with systemic steroids (initial dose: 1 mg/kg body weight per day - tapered to 5 mg/day) was initiated. Under this treatment, central serous detachment and panuveitis improved. The ophthalmological findings remained stable for half a year (Figure 1E and Figure 1F). During the following year the patient presented twice with recurrent ocular inflammation of the anterior and posterior segment with AC cells, typical granulomatous retrocorneal precipitates and macular edema, which improved after augmentation of the oral and local steroid therapy. Long term low-dose steroid therapy (5 mg prednisolone daily) was also administered due to recurrent cutaneous discomfort (Figure 2). Because of the typical appearance of the recurrent skin lesions - a yellowish-brown papulosquameous cicatricial exanthema - and because of improvement during prednisolone treatment we excluded the differential diagnosis of skin lesions caused by corticosteroids, which would appear as erythematous papulopustules. The patient developed latent diabetes mellitus under long term oral steroid therapy and additional immunosuppressive therapy with azathioprine (3 × 50 mg/day) was initiated to reduce the steroid dose. Due to side effects, azathioprine had to be stopped and therapy with methotrexate was commenced (20 mg/week per os). A 6-month follow-up examination showed stable findings, with no evidence of intraocular inflammation and subretinal fluid.
.
Figure 1: (A-B) Fundus photographs of both eyes with typical depigmentated retinochoroidal lesions after an acute panuveitic phase "sunset glow fundus" on (A) the right eye and (B) the left eye. (C-F) Imaging with OCT of both eyes: at first presentation with a serous bullous retinal detachment on (C) the right eye and (D) the left eye, and after 6-months of combined systemic steroidal and immunosuppressive therapy showing no evidence of subretinal fluid on (E) the right eye and (F) the left eye.
View Figure 1
.
Figure 2: Papulo-squameous cicatricial skin lesions in the lower back of the patient under non-inflammatory conditions.
View Figure 2
Discussion
Previous publications have reported other immunologically mediated diseases occurring together with Vogt-Koyanagi-Harada-syndrome, such as systemic sclerosis, Graves’ disease, IgA-Nephropathy, polyglandular autoimmune syndrome type I, recurrent polychondritis, aortitis, Guillain-Barré-syndrome and Diabetes mellitus [2,6]. The clinical course of ocular manifestations in Vogt-Koyanagi-Harada syndrome is quite varied. Our patient presented initially with bilateral panuveitis with optic disc edema and central serous retinal detachment with moderate anterior chamber cells and untypically non-granulomatous retrocorneal precipitates. In similarity to our findings VKH patients are described in the literature, who revealed the typical involvement of the posterior segment in combination with moderate anterior uveitis and non-granulomatous retrocorneal precipitates [7]. Interestingly, later on while developing recurrent stage, these patients disclosed granulomatous retrocorneal precipitates and a "sunset glow fundus" with Dalen-Fuchs nodules [7], corresponding to the subsequent findings in our patient (Figure 1A and Figure B).
This case report represents the first description of VKH and Morbus Darier occurring concurrently. The development of Morbus Darier is caused by a mutation on chromosome 12q24.1 in the ATP2A2 gene which encodes the sarco- and endoplasmic reticulum calcium-ATPase 2 (SERCA2), leading to dysfunction in the differentiation of keratinocytes [8,9]. The cause of VKH is not yet fully understood, however, there seem to be both autoimmune and infectious aspects. There is strong association of VKH to a variant of the HLA DRB1*0405 which leads to alteration of the MHC II - complex structure and changes antigen-presentation to CD4+ T cells. In this case, melanocytes are targets for destruction, which is induced by cytokines and chemokines secreted by B cells and CD4+ T cells in various tissue types such as skin, inner ear, meninges and uvea. Another study has shown that viral infection can also be involved in disease reactivation [10].
Tissue necrosis is the underlying pathological mechanism in Morbus Darier, which leads to the release of melanin. This suggests that Morbus Darier and VKH may exacerbate each other via inflammatory mechanisms.
Acknowledgements
The authors have no conflict of interest and have full control of all primary data. This work was presented as a poster on the conference of German Society of Ophthalmologists in Berlin in September 2014.
References
-
Sakata VM, da Silva FT, Hirata CE, de Carvalho JF, Yamamoto JH (2014) Diagnosis and classification of Vogt-Koyanagi-Harada disease. Autoimmunity reviews 13: 550-555.
-
Mota LA, Santos AB (2010) Vogt-Koyanagi-Harada's syndrome and its multisystem involvement. Rev Assoc Med Bras 56: 590-595.
-
Sato A, Anton-Lamprecht I, Schnyder UW (1977) Ultrastructure of dyskeratosis in morbus Darier. J Cutan Pathol 4: 173-184.
-
Damico FM, Kiss S, Young LH (2005) Vogt-Koyanagi-Harada disease. Semin Ophthalmol 20: 183-190.
-
Read RW, Holland GN, Rao NA, Tabbara KF, Ohno S, et al. (2001) Revised diagnostic criteria for Vogt-Koyanagi-Harada disease: report of an international committee on nomenclature. Am J Ophthalmol 131: 647-652.
-
Cavalli C, Gobbi PG, Colombo R, Riccardi A, Gorini M, et al. (1985) A case of scleroderma with sclerodermic renal crisis and association with the Vogt-Koyanagi-Harada syndrome. Presse Med 14: 1131-1134.
-
Yang P, Ren Y, Li B, Fang W, Meng Q, et al. (2007) Clinical characteristics of Vogt-Koyanagi-Harada syndrome in Chinese patients. Ophthalmology 114: 606-614.
-
Green EK, Gordon-Smith K, Burge SM, Grozeva D, Munro CS, et al. (2013) Novel ATP2A2 mutations in a large sample of individuals with Darier disease. J Dermatol 40: 259-266.
-
Sakuntabhai A, Ruiz-Perez V, Carter S, Jacobsen N, Burge S, et al. (1999) Mutations in ATP2A2, encoding a Ca2+ pump, cause Darier disease. Nat Genet 21: 271-277.
-
Hayasaka Y, Hayasaka S (2004) Almost simultaneous onset of Vogt-Koyanagi-Harada syndrome in co-workers, friends, and neighbors. Graefe's archive for clinical and experimental 242: 611-613.
