

Journal of Rheumatic Diseases and Treatment
Erdheim-Chester Disease: A Case of Cardiac and Hypophyseal Involvement Showing Positive Response with High Dose Pegylated Interferon-Α
Annie HN Law* and SI Yeo
Department of Rheumatology and Immunology, Singapore General Hospital, Singapore
*Corresponding author: Dr. Annie HN Law, Department of Rheumatology and Immunology, Academia Building Level 4, College Road, 169856, Singapore, Tel/Fax: 6321-4028/62207765, E-mail: annie.law.h.n@sgh.com.sg
J Rheum Dis Treat, JRDT-2-031, (Volume 2, Issue 1), Case Report; ISSN: 2469-5726
Received: November 30, 2015 | Accepted: February 05, 2016 | Published: February 07, 2016
Citation: Law AHN, Yeo SI (2016) Erdheim-Chester Disease: A Case of Cardiac and Hypophyseal Involvement Showing Positive Response with High Dose Pegylated Interferon-Α. J Rheum Dis Treat 2:031. 10.23937/2469-5726/1510031
Copyright: © 2016 Law AHN, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Erdheim-Chester disease (ECD) is a rare form of non-Langerhans cell histiocytosis, with multifaceted clinical presentations. There are currently fewer than 550 cases reported in the literature. This case illustrates the protean manifestations of ECD with multisystem involvement, including skeletal and extra skeletal (cutaneous, cardiac, pituitary, retroperitoneal, and pulmonary) manifestations. It reports on the complexity in making the diagnosis and subsequent positive response of ECD to pegylated interferon α.
Keywords
Erdheim Chester disease, Cardiac, Neurological, Interferon
Introduction
Erdheim-Chester disease (ECD) was first described by Jakob Erdheim’s pupil William Chester in 1930 [1]. It is a rare multisystemic, non-Langerhans histiocytic disorder derived from the monocyte-macrophage lineage. These histiocytes infiltrate tissues forming sheets of foamy histiocytes with surrounding inflammatory cells and fibrosis. The non-Langerhans cell express surface markers of CD 68 and do not express CD1a or S100 found in Langerhans cell histiocytosis. Typically tissue from ECD patients have CD 68+ CD1a – S100- expression [2]. ECD mainly affects men with a mean age of 55 years [3]. A delay in diagnosis of months to years is not unusual [4]. We describe a case with extensive multisystem involvement including cardiac and endocrine manifestations, and its response to high dose pegylated interferon (INF)-α.
Case Report
A 55-year-old gentleman was admitted to Singapore General Hospital in June 2013 with a one-year history of shortness of breath, bilateral knee pain, and weight loss. Examination revealed upper and lower eyelid xanthelasmata, bilateral gynecomastia, bibasal fine crepitations, hepatomegaly, and lower limb edema up to mid shin bilaterally. Cardiovascular examination was unremarkable.
Laboratory investigations performed were as follows: hemoglobin 11.5 g/dl; erythrocyte sedimentation rate (ESR) 100 mm/hr; C-reactive protein 53 mg/L; albumin 29 g/L; alanine aminotransferase 24 U/L; aspartate aminotransferase 29 U/L; testosterone 2.1 nmol/L (7.3-27.3 nmol/L); follicular stimulating hormone 0.4 U/L (1.2-8.1 U/L); luteinizing hormone 0.1 U/L (2-10.9 U/L); prolactin 51.5 UG/L (5-27.7 UG/L); oestradiol 188 pmol/L (44-186 pmol/L); beta human chorionic gonadotropin < 0.6 U/L (0-7.9U/L); synacthen test: cortisol at 0 min 140 nmol/L; cortisol (at 30 mins) 363 nmol/L; cortisol (at 60 mins) 323 nmol/L; antinuclear antibody 1:200 midbody; total cholesterol 3.29 mmol/L; triglycerides 1.47 mmol/L; high-density lipoprotein 0.47 mmol/L; low-density lipoprotein 2.15 mmol/L. A myeloma screen showed no monoclonal band, and free light chain kappa/lambda ratio was 1.5 (normal limits).
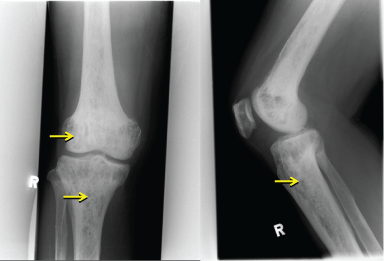
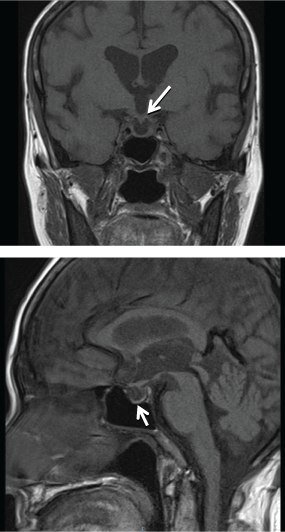
Radiography of the right knee (Figure 1) showed diffuse increased bone density with lytic lesions in the tibia and lower femur. Technetium (Tc) 99 m bone scintigraphy showed multiple lytic lesions in the distal femur, tibia and distal humerus, radius, ulnar and skull. Magnetic resonance imaging (MRI) of the pituitary gland was performed to determine the etiology of hypogonadotrophin hypogonadism and hyperprolactinemia, and showed that the pituitary gland was thinned out and flattened at the floor of the pituitary fossa with presence of pituitary stalk, consistent with an empty sella syndrome (Figure 2). Computed tomography (CT) scans of the thorax, abdomen, and pelvis showed a right pulmonary mass interposed between the middle and lower lobes and hepatomegaly. A CT guided biopsy of the right pulmonary mass revealed fibrous tissue with scattered chronic inflammatory cells and absence of malignant cells.
.
Figure 1: Right Knee X-ray: Lytic lesions over the metaphyses of the femur and tibia.
View Figure 1
.
Figure 2: T1 weighted image of empty sella - thinned out pituitary gland (white arrows).
View Figure 2
Positron emission tomography (PET)-CT scans revealed extensive symmetrical fluorodeoxyglucose (FDG) uptake at the diametaphyseal regions of both upper and lower extremities and coarse trabecular pattern in both humeral heads, right clavicular head, greater trochanters of both femurs, bilateral femoral condyles, tibiae, fibulae, calcanei, and tarsal bones. There was also presence of soft tissue thickening encasing the pericardium, thoracic aorta, coronary arteries including the left main, left circumflex and right coronary arteries, left kidney and left renal artery and vein, retroperitoneum, and around the superior mesenteric artery. There was no FDG avid abdominopelvic lymphadenopathy, focal hypermetabolic lesion seen in the liver, spleen, pancreas, gallbladder or adrenals. Pulmonary involvement was present, showing few small volume mildly FDG avid mediastinal lymph nodes in the prevascular and pericarinal stations and marked pleural and interstitial thickening throughout the lung fields with nodular mass-like consolidation along the right inferior oblique fissure. The imaged head and neck showed physiological tracer uptake in the brain, small volume bilateral cervical nodes with mild FDG uptake which was probably reactive in nature. The nasopharynx, or oropharynx, hypopharynx and thyroid gland appear unremarkable. These findings are illustrated on table 1.
.
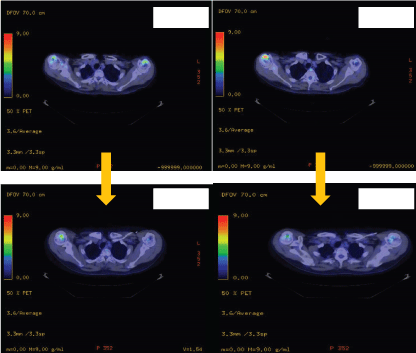
Figure 3a: Positron emission tomography showing reduce FDG uptake on the distal humeri post treatment.
View Figure 3a
.
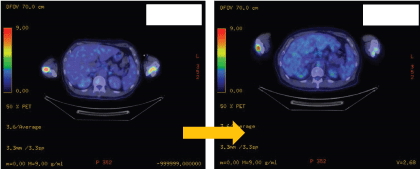
Figure 3b: Positron emission tomography showing reduce FDG uptake on the distal femur post treatment.
View Figure 3b
![]()
Table 1: Comparisons of FDG uptake on skeletal and cardiac involvement pre-treatment vs post-treatment.
View Table 1
A CT guided biopsy of the right supracondylar femur showed intertrabecular fibrosis with small clusters of CD 163 and CD 68 immunopositive foamy macrophages. No giant cells, eosinophils, or features of Langerhans cell histiocytosis were present. S100, CD 1a immunostaining were negative, and BRAF v600e was positive. These features were consistent with ECD. A two-dimensional echocardiography performed showed a normal left ventricular ejection fraction of 70% with a right atrial mass of 2.5 × 1.3 cm at the posterior wall and another at the lateral wall, which was not visualized on the PET-CT performed. During the course of his stay, a pacemaker was inserted as he developed ventricular fibrillation and bradycardia with sinus pause associated with multiple episodes of syncope.
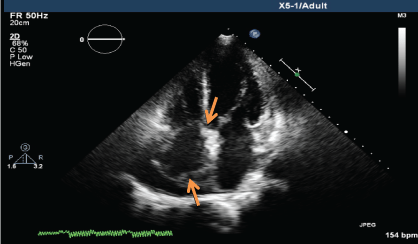
.
Figure 4a: Transthoracic echocardiogram (TTE) showing mass lesion in the right atrium close to the tricuspid valve and at the posterior wall (June 2013).
View Figure 4a
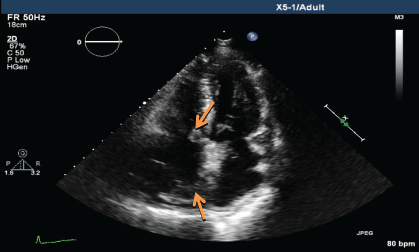
.
Figure 4b: TTE showing persistent mass lesion in the right atrium close to the tricuspid valve and at the posterior wall (March 2014).
View Figure 4b
Upon histopathological confirmation of ECD, the patient was given subcutaneous peginterferon-α of 180 mcg once/week and prednisolone of 1 mg/kg/day. After 3 months of treatment, his shortness of breath and joint pain resolved. Repeat laboratory test showed declining ESR of 29 mm/hr and a repeat PET-CT scan showed significant improvement in pericardial soft tissue thickening encasing the thoracic aorta and coronary arteries, with skeletal lesions also showing quantitative reduction of the metabolic activity (Figure 3a and Figure 3b) and no new hypermetabolic lesions. (Table 1). A repeated two- dimensional echocardiogram 18 months later showed regression of the right atrial mass (Figure 4a, Figure 4b, Figure 4c and Figure 4d).
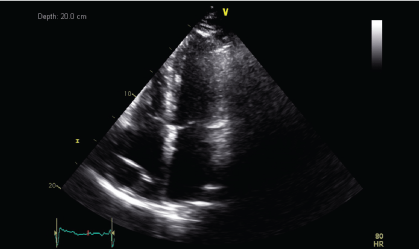
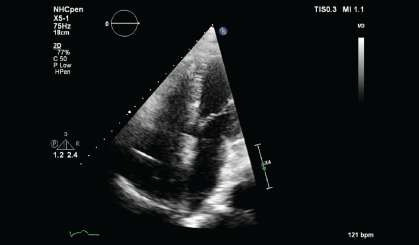
Discussion
ECD is a rare multisystem disease of unknown pathogenesis [5]. The most common site of infiltration is at the long bones, which are found in 96% of patients with ECD, of whom up to 50% suffer from bone pain [4]. Extraskeletal manifestations are not unusual as the reported frequency of cardiac involvement is as high as 70%, central nervous system 15-25%, renal involvement due to pseudoretroperitoneal fibrosis up to 30%, pulmonary 22%, exophthalmos 27%, and mucocutaneous involvement 28%, respectively [6-8]. A definite diagnosis of ECD is reliant on histological and radiological confirmation. In our patient, this was confirmed on tissue biopsy which showed infiltration by foamy histiocytes nested among polymorphic granuloma and fibrosis or xanthogranulomatosis with immunostaining of CD 68+ and CD1a-. The diagnosis was further supported by strong uptake of the distal ends of femur and humerus on Tc99m bone scintigraphy. The use of bone scintigraphy is essential in diagnosing ECD as it provides an overview on the site of skeletal involvement and enable biopsy to be performed at the optimal site. In recent years, it has been considered as a possible diagnostic criterion for ECD as majority of the patients with ECD fulfils the typical skeletal findings of symmetrical and increase uptake on the diaphyseal and metaphyseal regions of the long bone [7].
This case illustrates the unique endocrine presentation with empty sella syndrome and involvement of anterior pituitary, sparing the posterior lobe. Although central diabetes insipidus has been reported to be the most frequent endocrine manifestation seen in ECD, this was not the case in our patient [9]. To date, only one other case describes an ECD patient with empty sella syndrome [10]. Recently, earlier recognition of ECD and treatment has improved the mortality rates of the disease. In a series of patients studied in 2004, cardiovascular involvement carries a poor prognosis with mortality as high as 60% and a mean survival of 19.2 months after diagnosis [11] while a recent study in 2011 showed a more favorable outcome in patient with CNS involvement with mortality rate of 26% and five-year survival rate of 68% after treatment with IFN-α [3].
We chose to treat with prednisolone 1 mg/kg/day as it better controls the symptoms of the disease [12]. However, an observational cohort study done by Arnaud et al. concluded that there was no survival benefit in patients treated with corticosteroids [3]. High dose pegylated IFN-α of 180 mcg/week was started as opposed to ‘standard’ low dose IFN-α of 3 mIU 3x/week as Haroche et al. concluded that standard dose IFN-α was ineffective [13]. The efficacy of high dose IFN-α was confirmed by Hervier et al. who followed disease progression of 24 patients who received high dose IFN-α ≥ 18 mIU/wk or pegylated IFN-α ≥ 180 mcg/wk. Disease improvement was defined by combined morphological and clinical improvement. Morphological improvement was defined as reduction of tissue infiltration and decreased FDG-PET uptake, while clinical improvement was defined as a decrease in cutaneous and orbital infiltration, pelvic, and bone pain, respectively. Stabilization of disease was defined by a lack of change in clinical and morphological entity [14]. The study concluded that 16 patients (67%) had improvement (46%) and stabilization (21%) of the disease. Of those patients, pituitary and cardiac improvement was observed in 29% of them [14]. Similarly, treatment with high dose pegylated IFN-α resulted in improvement of osteosclerotic lesions in the long bones and pericardial soft tissue thickening with normalization of ESR (18 mm/hr) in our patient. He did not suffer any side effects of asthenia, myalgia, pruritus, thrombocytopenia, and depression from the treatment.
Another promising treatment for patients with refractory ECD is the inhibitor of BRAF v600e mutant, vemurafenib. BRAF is a proto-oncogene which directs cell growth. Mutation of BRAF is related to tumors with abnormal cell growth [15]. BRAF v600e mutations were identified in 51-54% of the tissue samples of patients with ECD via pyrosequencing [16,17]. However, using different techniques, Cangi and colleagues found that all of their 18 ECD patients had positive BRAF v600e mutation in peripheral blood samples and biopsies with the use of locked nucleic acid-pyrosequencing technique [18]. These findings were further confirmed by use of droplet digital polymerase chain reaction detection of BRAF v600e [18]. Therefore, they hypothesized that potentially all patients with ECD might have the BRAF v600e mutation. Traditionally, vemurafenib is used in patients with melanoma and hairy cell leukemia as it has been shown to improve survival in these diseases [19]. Haroche et al. treated three ECD patients refractory to IFN-α with vemurafenib, with significant clinical and radiological (PET-CT) improvement at one month and four months post-treatment [20]. The dose administered was 1920 mg/day and tapered to 960 mg/day from day 30. All the patients treated suffered cutaneous reaction with erythema, pruritus, and keratosis pilaris, thereby limiting prolonged use at high dose [21]. Nonetheless, it remains a promising alternative in patients with life threatening disease who are refractory to standard treatment. Other alternatives include systemic chemotherapy with vinblastine and etoposide, which are agents used in treatment of Langerhans cell histiocytosis [21]. Use of anakinra (interleukin 1 inhibitor), [22] imatinib, [23] cladribine, [24,25] cyclopshosphamide, [26] and infliximab, [27] have also been reported.
There are several challenges in identifying disease severity and progression in ECD radiologically and serologically. The reason is because there are limited data on what is the “ideal” modality for radiological assessment and there is no single biomarker to assess severity or progression. A recent study published by Garcia-Gomez et al. suggested the use of bone scintigraphy and PET-CT at baseline and follow-up. At baseline, these modalities help to locate site of involvement for optimal biopsy and evaluates the extension of disease while at follow-up, it helps to evaluate response to treatment and monitor disease involvement [28]. Nevertheless, PET-CT has its limitations when assessing organ specific disease as sensitivity can varies from 4.3% to 78.3% on the organ studied while maintaining high specificity [29]. For instance, assessment of cardiac disease with cardiac MRI versus PET-CT yielded a specificity of 100% and sensitivity of 9.1% with a positive predictive value of 100% and negative predictive value of 28.6% at diagnosis and at follow-up [29]. The use of PET-CT showed a much lower sensitivity when compared to cardiac MRI as pseudotumours of the heart are often masked by physiological uptake present in the myocardium [29]. Therefore, PET-CT may serve to be a better modality for follow-up use than initial assessment due to the low sensitivities. However, its role as a tool for initial assessment and follow-up should be considered as it provides a global assessment of the extent and severity of the disease. In our case, PET-CT was chosen as the follow-up modality as our patient has a pacemaker that is MRI incompatible.
There is no specific serological marker in identifying disease activity or severity. C-reactive protein has been used as a surrogate marker for this purpose [7]. Specific markers could be identified if the underlying pathogenesis of the disease and the downstream cytokines produced by mutated and non-mutated histiocytes can be elucidated. The use of LNA-pyrosequencing DNA technique in detecting patients with BRAF v600e mutations at diagnosis may prove to be a rapid, sensitive, and specific diagnostic tool in ECD. Further prospective studies are needed for validation of this test so that we can better understand if BRAF v600e mutation may be the pathogenic culprit of ECD. This will enable targeted treatment and improved outcomes.
Conclusion
ECD is a complex multi-system disease that is difficult to diagnose. To date, there is no proven cure for the disease and effective treatments are lacking. We need to further understand disease pathogenesis and the natural course of the disease so that we can diagnose early, prognosticate, and improve treatment in this group of patients.
References
-
Chester W (1930) Über lipoidgranulomatose. Virchows Archiv für pathologische Anatomie und Physiologie und für klinische Medizin 279: 561-602.
-
Dagna L, Girlanda S, Langheim S, Rizzo N, Bozzolo EP, et al. (2010) Erdheim-Chester disease: report on a case and new insights on its immunopathogenesis. Rheumatology (Oxford) 49: 1203-1206.
-
Arnaud L, Hervier B, Néel A, Hamidou MA, Kahn JE, et al. (2011) CNS involvement and treatment with interferon-α are independent prognostic factors in Erdheim-Chester disease: a multicenter survival analysis of 53 patients. Blood 117: 2778-2782.
-
Haroche J, Amoura Z, Wechsler B, Veyssier-Belot C, Charlotte F, et al. (2007) Erdheim-Chester disease. Presse Med 36: 1663-1668.
-
Cavalli G, Guglielmi B, Berti A, Campochiaro C, Sabbadini MG, et al. (2013) The multifaceted clinical presentations and manifestations of Erdheim-Chester disease: comprehensive review of the literature and of 10 new cases. Ann Rheum Dis 72: 1691-1695.
-
Haroche J, Cluzel P, Toledano D, Montalescot G, Touitou D, et al. (2009) Images in cardiovascular medicine. Cardiac involvement in Erdheim-Chester disease: magnetic resonance and computed tomographics scan imaging in a monocentric series of 37 patients. Circulation 119: e597-598.
-
Haroche J, Arnaud L, Cohen-Aubart F, Hervier B, Charlotte F, et al. (2013) Erdheim-Chester disease. Rheum Dis Clin North Am 39: 299-311.
-
Veyssier-Belot C, Cacoub P, Caparros-Lefebvre D, Wechsler J, Brun B, et al. (1996) Erdheim-Chester disease. Clinical and radiologic characteristics of 59 cases. Medicine (Baltimore) 75: 157-169.
-
Manaka K, Makita N, Iiri T (2014) Erdheim-Chester disease and pituitary involvement: a unique case and the literature. Endocr J 61: 185-194.
-
Tien R, Kucharczyk J, Newton TH, Citron JT, Duffy TJ (1990) MR of diabetes insipidus in a patient with Erdheim-Chester disease: case report. AJNR Am J Neuroradiol 11: 1267-1270.
-
Haroche J, Amoura Z, Dion E, Wechsler B, Costedoat-Chalumeau N, et al. (2004) Cardiovascular involvement, an overlooked feature of Erdheim-Chester disease: report of 6 new cases and a literature review. Medicine (Baltimore) 83: 371-392.
-
Mazor RD, Mazor MM, Shoenfeld Y (2013) Strategies and treatment alternatives in the management of Erdheim-Chester disease. Expert Opin Orphan Drug 11: 891-899
-
Haroche J, Amoura Z, Trad SG, Wechsler B, Cluzel P, et al. (2006) Variability in the efficacy of interferon-alpha in Erdheim-Chester disease by patient and site of involvement: results in eight patients. Arthritis Rheum 54: 3330-3336.
-
Hervier B, Arnaud L, Charlotte F, Wechsler B, Piette JC, et al. (2012) Treatment of Erdheim-Chester disease with long-term high-dose interferon-α. Semin Arthritis Rheum 41: 907-913
-
Davies H, Bignell GR, Cox C, Stephens P, Edkins S, et al. (2002) Mutations of the BRAF gene in human cancer. Nature 417: 949-954.
-
Emile JF, Charlotte F, Amoura Z, Haroche J (2013) BRAF mutations in Erdheim-Chester disease. J Clin Oncol 31: 398.
-
Haroche J, Charlotte F, Arnaud L, von Deimling A, Hélias-Rodzewicz Z, et al. (2012) High prevalence of BRAF V600E mutations in Erdheim-Chester disease but not in other non-Langerhans cell histiocytoses. Blood 120: 2700-2703.
-
Cangi MG, Biavasco R, Cavalli G, Grassini G, Dal-Cin E, et al. (2015) BRAFV600E-mutation is invariably present and associated to oncogene-induced senescence in Erdheim-Chester disease. Ann Rheum Dis 74: 1596-1602.
-
Dietrich S, Glimm H, Andrulis M, von Kalle C, Ho AD, et al. (2012) BRAF inhibition in refractory hairy-cell leukemia. N Engl J Med 366: 2038-2040.
-
Haroche J, Cohen-Aubart F, Emile JF, Arnaud L, Maksud P, et al. (2013) Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim-Chester disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood 121: 1495-1500.
-
Gadner H, Grois N, Pötschger U, Minkov M, Aricò M, et al. (2008) Improved outcome in multisystem Langerhans cell histiocytosis is associated with therapy intensification. Blood 111: 2556-2562.
-
Aouba A, Georgin-Lavialle S, Pagnoux C, Martin Silva N, Renand A, et al. (2010) Rationale and efficacy of interleukin-1 targeting in Erdheim-Chester disease. Blood 116: 4070-4076.
-
Janku F, Amin HM, Yang D, Garrido-Laguna I, Trent JC, et al. (2010) Response of histiocytoses to imatinib mesylate: fire to ashes. J Clin Oncol 28: e633-636.
-
Myra C, Sloper L, Tighe PJ, McIntosh RS, Stevens SE, et al. (2004) Treatment of Erdheim-Chester disease with cladribine: a rational approach. Br J Ophthalmol 88: 844-847.
-
Adam Z, Řehák Z, Koukalová R, Bortlíček Z, Krejčí M, et al. (2014) [PET-CT documented complete remission of Erdheim-Chester disease, lasting more than 4 years from treatment initiation with cladribine. Vnitr Lek 60: 499, 501-511.
-
Bourke SC, Nicholson AG, Gibson GJ (2003) Erdheim-Chester disease: pulmonary infiltration responding to cyclophosphamide and prednisolone. Thorax 58: 1004-1005.
-
Dagna L, Corti A, Langheim S, Guglielmi B, De Cobelli F, et al. (2012) Tumor necrosis factor α as a master regulator of inflammation in Erdheim-Chester disease: rationale for the treatment of patients with infliximab. J Clin Oncol 30: e286-290.
-
García-Gómez FJ, Acevedo-Báñez I, Martínez-Castillo R, Tirado-Hospital JL, Cuenca-Cuenca JI, et al. (2015) The role of 18FDG, 18FDOPA PET/CT and 99mTc bone scintigraphy imaging in Erdheim-Chester disease. Eur J Radiol 84: 1586-1592.
-
Arnaud L, Malek Z, Archambaud F, Kas A, Toledano D, et al. (2009) 18F-fluorodeoxyglucose-positron emission tomography scanning is more useful in follow-up than in the initial assessment of patients with Erdheim-Chester disease. Arthritis Rheum 60: 3128-3138.
