

Journal of Genetics and Genome Research
A Comparison of SNaPshot Minisequencing and HRM Analysis in mtSNP Genotyping with Reference Samples from East Timor
Luis Souto1*, Filipa Tavares1, Helena Moreira1 and Fatima Pinheiro2,3
1Department of Biology, University of Aveiro, Aveiro, Portugal
2ICBAS, University of Porto, Porto, Portugal
3Medicine Faculty, University of Porto, Porto, Portugal
*Corresponding author:
Luis Souto, Department of Biology, University of Aveiro, Aveiro, Portugal, Tel: (+351) 234 370 975, Fax: (+351) 234 372 587, E-mail: lsouto@ua.pt
J Genet Genome Res, JGGR-2-021, (Volume 2, Issue 2), Research Article; ISSN: 2378-3648
Received: October 01, 2015 | Accepted: December 23, 2015 | Published: December 28, 2015
Citation: Souto L, Tavares F, Moreira H, Pinheiro F (2015) A Comparison of SNaPshot Minisequencing and HRM Analysis in mtSNP Genotyping with Reference Samples from East Timor. J Genet Genome Res 2:021. 10.23937/2378-3648/1410021
Copyright: © 2015 Souto L, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
The analysis of mtDNA in forensic samples is commonly carried out by direct sequencing of the hyper variable regions of the control region, although with limited power of discrimination. Genotyping SNPs in the coding region of mtDNA can provide additional information and increase the discrimination power of mtDNA typing. In this study, we compare two methodologies for the detection of 9 SNPs in the control region of mtDNA: SNaPshot minisequencing and Real Time PCR, through High-Resolution Melting (HRM) analysis. The selected markers were used in 36 blood samples collected from East Timor volunteers. Results showed that SNaPshot is a more precise, robust and reliable methodology than HRM analysis. However, HRM analysis has the potential to function as a rapid and inexpensive pre-screening method for forensic samples prior to minisequencing.
Keywords
East Timor, HRM analysis, Minisequencing, mtDNA, SNPs
Introduction
Human mitochondrial DNA (mtDNA) is a useful marker for studying maternal biogeography ancestry, and is widely used in forensic casework and human population studies due to its specific features, namely: rapid mutation rate, high copy number per cell, lack of recombination and maternal inheritance [1-3].
The maternal inheritance of mtDNA determines the evolution of the molecule along a phylogeny. This mode of inheritance results in a natural grouping of sequence haplotypes into clusters, referred to as haplogroups (Hg), which are named with an alternating order of letters and number in descending hierarchical order. The members of a haplogroup carry a specific sequence motif as a consequence of sharing a common ancestor [4].
This type of phylogeographic information can be useful in the forensic context when the geographic origin of an unknown sample is under investigation [5,6].
Most of the mtDNA sequence variations are located in the control region hypervariable segments I and II (HVS-I and HVS-II). Sequencing HVS-I and HVS-II is the most common method for mtDNA analysis in forensic identification, despite its limited power of discrimination [7]. The analysis of coding region mtDNA single nucleotide polymorphisms (mtSNPs) increased in recent years, with multiple strategies proposed to facilitate mtDNA SNP typing [8]. The recent developments in whole genome "next generation" technologies [9], while being able to sequencing entire mitochondrial genomes with unprecedented capabilities, is still not feasible as a routine approach available to a common forensic genetics lab.
Herein, we compare two methodologies: SNaPshot minisequencing and High-Resolution Melting (HRM) analysis in detecting mtDNA SNPs.
The SNaPshot™ reaction is a popular standardized minisequencing tool for genotyping mtSNPs among forensic labs [10,11]. HRM analysis is a post-PCR analysis used to identify genetic variations in a fragment of real-time PCR amplified DNA by comparing fluorescence signals as function of melting temperatures. Alleles producing distinct melting curves can be compared with reference samples [12].
HRM analysis has been considerably widespread as a screening tool for several pathological conditions [13,14]. In forensic samples, HRM, while already suggested as pre-screening method [15] it is still scarcely used as an application in forensic genetics, particularly in mtSNPs genotyping.
Materials and Methods
DNA samples
In the frame of a genetic research project on the East Timor population [16,17], a total of 36 blood samples were collected from East Timorese university students volunteers, following informed consent statements. From each volunteer, 1mL of blood was collected.
Genomic DNA was extracted from whole blood using the Chelex®-100 modified method [18].
SNPs and Primer selection
The SNPs used in this study were selected from the Asian mitochondrial tree, previously described in Alvarez-Iglesias et al [19].
Primers were chosen in order to meet both Minisequencing and HRM analysis criteria; in particularly they're expected to detect a single SNP. Primer and amplicon details are referred in table 1.
![]()
Table 1: SNP coding region, primer sequences and final concentration for PCR amplification (according to Alvarez-Iglesias et al [19]).
View Table 1
Positive and negative (wild type) SNP controls were used during the amplification and genotyping processes in both techniques.
Minisequencing
PCR-multiplex amplification and purification: Multiplex PCR amplification was carried out in a total volume of 15 μl, containing 1x QIAGEN Multiplex PCR Master Mix (Qiagen), PCR primers concentrations as specified in table 1 and 1 μl of DNA.
Thermal cycling was carried out on Icycler (Bio-Rad), with the following conditions: 95°C pre-incubation step for 15 min; 30 cycles of 94°C denaturation for 30 sec, annealing at 60°C for 90 sec and extension at 72°C during 90 sec; followed by 15 min for final extension at 72°C and 4°C until removed from the thermocycler. PCR products and negative controls were checked by agarose gel electrophoresis.
PCR products were purified to remove excess of primers and unincorporated dNTPs by adding 2 μl of ExoSAP-IT (USB Corporation) to each 5 μl PCR product. Reactions were incubated at 37°C for 15 min followed by 80°C for 15 min for enzyme deactivation.
Multiplex Minisequencing using SNaPshot and purification: The minisequencing reaction was performed using SNaPshot™ Kit (Applied Biosystems). Extension primers range between 25 and 67 bp modified with 5' end non-homologous tails poly (dC) or poly (dGACT) (Table 2).
![]()
Table 2: SNP coding region, primer extension sequences and final concentration for minisequencing reaction (according to Alvarez-Iglesias et al [19]).
View Table 2
The minisequencing reaction was carried out on Icycler (Bio-Rad Laboratories) using 3.5 μl of the SNaPshot™ Multiplex Ready Reaction Mix (Applied Biosystems), 1.5 μl purified PCR product, and 1.5 μl of extension primers mix in a final volume of 10 μl. The PCR conditions were: 25 cycles of denaturation at 96°C during 10 sec, annealing at 50°C for 5 sec and extension at 60°C during 30 sec. To remove the unincorporated ddNTPs, the final product was treated with 1 μl of Shrimp alkaline phosphatase (SAP) (Promega Corporation) at 37°C for 60 min followed by 80°C for 15 min for enzyme inactivation.
Electrophoresis detection: Minisequencing products (1 μl) were mixed with 10 μl of Hi-Di™ Formamide and 1 μl of GeneScan-120 LIZ (both Applied Biosystems). Samples were denatured at 95°C for 5 min and quickly cooled on ice for 5 min. Electrophoresis was run on an ABI PRISM® 310 Genetic Analyzer (Applied Biosystems) using Performance Optimum Polymer 6 (POP-6®). Resulting data were analyzed using GeneMapper® ID Software v3.2 (Applied Biosystems).
Real-Time PCR: HRM analysis
Real-Time PCR reactions were carried in a fluorometer thermal cycler CFX96™ Real-Time PCR Detection System (Bio-Rad Laboratories) using 20 μl of total reaction volume containing 1x of SsoFast™ EvaGreen® Supermix (Bio-Rad Laboratories, Hercules, CA, USA), 0.3 μM of each forward and reverse primer (Table 1), and 1 μl of DNA. Real-Time PCR conditions: initial denaturation at 98°C for 2 min, followed by 40 cycles at 98°C for 5 sec and 30 sec at the primer's specific annealing temperature (Table 1), with collection of fluorescence signals at the end of each cycle. Data were collected and processed using the software Bio-Rad CFX Manager 2.0 (Bio-Rad Laboratories).
For HRM analysis, PCR products were denatured at 95°C for 1 min and then annealed at 50°C for 5 min in order to allow for correct annealing of DNA duplexes. These two steps were followed by melting curve ranging from 65°C to 95°C with temperature increments of 0.2°C every 10s. The fluorescence data were acquired by the end of each melting temperature.
Collected fluorescence data were processed using the Precision Melting Analysis™ Software 1.1 (Bio-Rad Laboratories) to generate melting curves as a function of temperature and difference curves for easier visual identification of clusters. Raw fluorescence data was subjected to normalization and temperature shifting in order to remove background fluorescence, assisting in visual interpretation and automatic grouping of similar melting curves.
When justified, Real-Time PCR amplified products with the correspondent primer are sent to external facilities for automated sequencing.
Results
The results obtained for the two SNP genotyping methodologies under study (SNaPshot and HRM analysis) can be found in table 3. Figure 1 represents the output files of consistent results for one of the studied SNP in the evaluated methodologies.
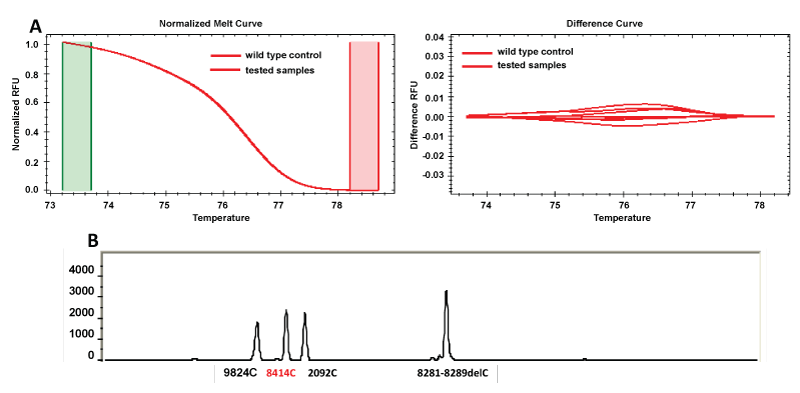
Figure 1: Representative output files of some consistent samples (1, 2, 5, 15 and 31) for SNP 8414 in the evaluated methodologies. (A) HRM results (normalized and difference curves); (B) SNaPshot readout after multiplex PCR.
View Figure 1
![]()
Table 3: Comparison of SNPs obtained in two compared methodologies [" " for wild type, which matches the corresponding base in the revised Cambridge Reference Sequence (rCRS) [20]; the nucleotides (A, T, C, G) specify the polymorphism detected].
View Table 3
When comparing the two methodologies, more than 99% of loci results were consistent. However, discordances (labeled as * in table 3) were found in samples 28, 49 and 62 for SNP 8414 (Figure 2). In these three samples, the HRM results demonstrate that the tested samples present differences regarding to the wild type control (which matches to the corresponding base in the revised Cambridge Reference Sequence (rCRS) [20]). However the SNaPshot result demonstrates that these samples do not present the SNP 8414, which means that we are in the presence of a wild type sample. The discordant results were confirmed by reamplification and sequencing the real-time PCR products. Figure 3 represents a sequence electropherogram that demonstrates the presence of a SNP (8440) different from the SNP under study (8414).
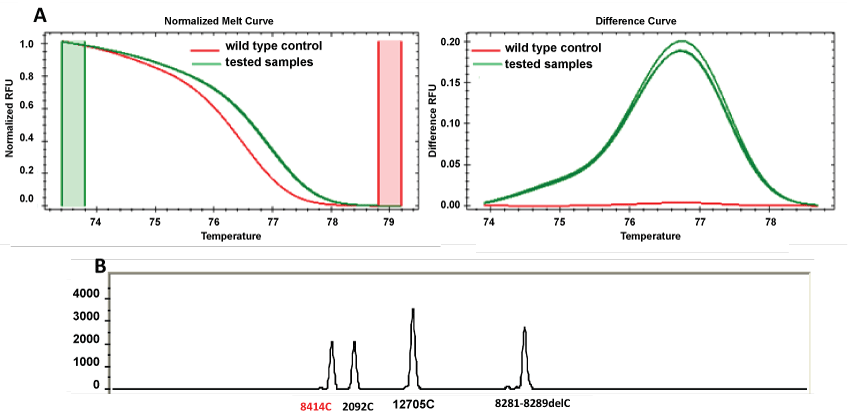
Figure 2: Representative output files of discordant samples (28, 49 and 62) for SNP 8414 in the evaluated methodologies. (A) HRM results (normalized and difference curves); (B) SNaPshot readout after multiplex PCR.
View Figure 2
Discussion
In SNP 8414, according to SNaPshot, samples 28, 49 and 62 were recorded as wild type while HRM analysis indicates a different melting curve from the wild type control sample (Figure 2).
Inconsistencies in SNP 8414 suggest a non-specificity of primers in HRM analysis. Phylogenetically, samples 28, 49 and 62 were classified as subhaplogroup R9c based on previous studies [17]. Consequently SNP 8414 is not phylogenetically expected to be present in subhaplogroup R9c, taking in account its specificity to haplogroup D4.
The comparison of primers used to detect SNP 8414 with the revised Cambridge Reference Sequence (rCRS) [20] confirms that another polymorphism, SNP 8440 (which is present in the haplogroup R9c1a), was found in the amplicon. As a result, the polymorphism detected with HRM analysis could be 8440 (which classifies the samples in the subhaplogroup R9c1a) and not SNP 8414. The presence of polymorphism 8440 in samples 28, 49 and 62 was confirmed by sequencing its real-time PCR products (Figure 3).
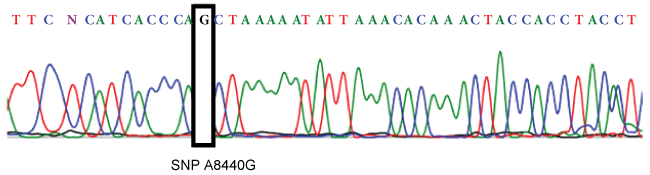
Figure 3: Sequence electropherogram for SNP 8440. The A nucleotide is wild type, which matches to the corresponding base in the revised Cambridge Reference Sequence (rCRS) [20], and the G nucleotide is the polymorphism detected.
View Figure 3
HRM analysis is based on the comparison of melting curves that have a particular shape according to the amplicon's nucleotide composition, without determining what polymorphism is present at a specific position. This particularity makes the HRM analysis disadvantageous since it does not identify the specific polymorphism in the analyzed amplicon.
On the other hand, SNaPshot is a method that uses extension primers that anneal to its target DNA immediately adjacent to the SNP under analysis. The fact that SNaPshot uses specific primers for each polymorphism makes this methodology more specific in the detection of SNPs when compared to HRM analysis. In addition, it should be noted that the detection method uses a common technology (Capillary electrophoresis) as in current STR analysis.
In order to improve the specificity of HRM analysis, it is important to use primers that amplify only one SNP per reaction. Occasionally, two or more SNPs may be located close to each other, making it difficult to design primers to locate a single SNP. However, it is possible to amplify a region that contains multiple SNPs and carry out HRM analysis, as long as control samples for each SNP under analysis are included in the protocol.
Analysis of mtDNA has proven to be a good alternative in forensic applications, when autosomal STR markers fail to give conclusive results due to the low amount of intact nuclear DNA.
A screening method of forensic samples based on SNP determination that could spare sequencing efforts on casework analysis is of relevant interest. In addition, SNPs increase the discrimination power of mtDNA control region sequencing.
In conclusion, considering that the PCR amplification is an essential part of the forensic sample analysis, HRM analysis may contribute to overcoming the low power of discrimination of mtDNA control region sequencing. HRM analysis can be integrated into laboratory routines as a method of sample screening, provided appropriate phylogenetic control samples are used and complemented by subsequent SNaPshot genotyping.
Acknowledgements
We would like to thank the East Timorese volunteers for their contribution in this study. Our work was supported by the FCT Project: PTDC/CS-ANT/108558/2008, Programa Operacional Temático, Fatores de Competitividade (COMPETE), Quadro Comunitário de Apoio III and FEDER.
References
-
Kaye N. Ballantyne, Mannis van Oven, Arwin Ralf, Mark Stoneking, R. John Mitchell, et al. (2011) MtDNA SNP multiplexes for efficient inference of matrilineal genetic ancestry within Oceania. Forensic Sci Int Genet 6: 425-436.
-
A Carracedo (2005) Forensic DNA typing protocols. Methods in Molecular Biology, pp 1-11.
-
Ingman M, Kaessmann H, Pääbo S, Gyllensten U (2000) Mitochondrial genome variation and the origin of modern humans. Nature 408: 708-713.
-
van Oven M, Kayser M (2009) Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum Mutat 30: E386-394.
-
Salas A, Bandelt HJ, Macaulay V, Richards MB (2007) Phylogeographic investigations: the role of trees in forensic genetics. Forensic Sci Int 168: 1-13.
-
Bandelt HJ, van Oven M, Salas A (2012) Haplogrouping mitochondrial DNA sequences in legal medicine/forensic genetics. Int J Legal Med 126: 901-916.
-
Barbosa AB, da Silva LA, Azevedo DA, Balbino VQ, Mauricio-da-Silva L (2008) Mitochondrial DNA control region polymorphism in the population of Alagoas state, north-eastern Brazil. Journal of Forensic Sciences, 53: 142-146.
-
Sobrino B, Brión M, Carracedo A (2005) SNPs in forensic genetics: a review on SNP typing methodologies. Forensic Sci Int 154: 181-194.
-
Parson W, Strobl C, Huber G, Zimmermann B, Gomes SM, et al. (2013) Evaluation of next generation mtGenome sequencing using the Ion Torrent Personal Genome Machine (PGM). Forensic Sci Int Genet 7: 543-549.
-
Brandstätter A, Parsons TJ, Parson W (2003) Rapid screening of mtDNA coding region SNPs for the identification of west European Caucasian haplogroups. Int J Legal Med 117: 291-298.
-
Quintáns B, Alvarez-Iglesias V, Salas A, Phillips C, Lareu MV, et al. (2004) Typing of mitochondrial DNA coding region SNPs of forensic and anthropological interest using SNaPshot minisequencing. Forensic Sci Int 140: 251-257.
-
Reed GH, Wittwer CT (2004) Sensitivity and specificity of single-nucleotide polymorphism scanning by high-resolution melting analysis. Clin Chem 50: 1748-1754.
-
Er TK, Chang JG (2012) High-resolution melting: applications in genetic disorders. Clin Chim Acta 414: 197-201.
-
Dobrowolski SF, Hendrickx AT, van den Bosch BJ, Smeets HJ, Gray J, et al. (2009) Identifying sequence variants in the human mitochondrial genome using high-resolution melt (HRM) profiling. Hum Mutat 30: 891-898.
-
Gidlöf O, Burvall S, Edvinsson L, Montelius M, Allen M, et al. (2009) Complete discrimination of six individuals based on high-resolution melting of hypervariable regions I and II of the mitochondrial genome. Biotechniques 47: 671-678.
-
Souto L, Gusmão L, Amorim A, Côrte-Real F, Vieira DN (2006) Y-STR haplotype diversity in distinct linguistic groups from East Timor. Am J Hum Biol 18: 691-701.
-
L. Souto, A.M. Rocha, A. Pires, E. Ferreira, M. Kayser, et al. (2006) Mitochondrial DNA variability in populations from East Timor (Timor Leste). International Congress Series, 1288: 115-117.
-
Walsh PS, Metzger DA, Higushi R (1991) Chelex-100 as a Medium for Simple Extraction of DNA for Pcr-Based Typing from Forensic Material, Biotechniques, 10: 506-513.
-
Alvarez-Iglesias V, Jaime JC, Carracedo A, Salas A (2007) Coding region mitochondrial DNA SNPs: targeting East Asian and Native American haplogroups. Forensic Sci Int Genet 1: 44-55.
-
Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, et al. (1999) Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet 23: 147.
