

Journal of Clinical Gastroenterology and Treatment
MMPs, ADAMs and Their Natural Inhibitors in Inflammatory Bowel Disease: Involvement of Oxidative Stress
Filippo Fontani1, Vladana Domazetovic1, Tommaso Marcucci2, Maria Teresa Vincenzini1 and Teresa Iantomasi1*
1Department of Biomedical, Experimental and Clinical Sciences "Mario Serio", University of Florence, Italy
2Santa Maria Annunziata Hospital, Section of General Surgery, Ponte a Niccheri (Florence), Italy
*Corresponding author:
Teresa Iantomasi, Department of Biomedical, Experimental and Clinical Sciences "Mario Serio", University of Florence, Viale Morgagni, 50, 50134 Firenze, Italy, Tel: 00390552751200, E-mail: tiantomasi@unifi.it
J Clin Gastroenterol Treat, JCGT-3-039, (Volume 3, Issue 1), Review Article; ISSN: 2469-584X
Received: October 26, 2016 | Accepted: January 03, 2017 | Published: January 05, 2017
Citation: Fontani F, Domazetovic V, Marcucci T, Vincenzini MT, Iantomasi T (2017) MMPs, ADAMs and Their Natural Inhibitors in Inflammatory Bowel Disease: Involvement of Oxidative Stress. J Clin Gastroenterol Treat 3:039.
Copyright: © 2017 Fontani F, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Matrix metalloproteinases (MMPs) are enzymes involved in the degradation of extracellular matrix proteins and in several functions such as wound repair. The regulation of MMPs occurs at various levels including expression and inhibition by tissue specific inhibitors of MMPs (TIMPs). An alteration of intracellular redox state can modulate the activation and/or expression of MMPs. The increased expression and activity of MMPs are related to inflammatory state and pathogenesis of inflammatory bowel disease (IBD). In particular, an imbalance between MMPs and TIMPs is present at intestinal level in IBD, and this can be responsible of alterations of different processes such as repair of damaged mucosa, function and migration of immune cells and mucosal ulceration. A disintegrin and metalloproteinase 17 (ADAM17), belonging to ADAM family, plays an important role in the intestinal inflammation and increases in human colonic mucosa of IBD patients. In this review we report the effects of MMP, ADAM17 and TIMP altered levels on damage amplification caused by inflammation and oxidative stress in IBD. Moreover, the review evidences the role of the intracellular redox state and antioxidants on regulation of expression and activity of some MMPs in condition of oxidative stress in IBD.
Keywords
MMPs, ADAM17, IBD, Oxidative stress, Antioxidants
Introduction
The inflammatory bowel disease (IBD) includes two major forms of chronic intestinal disorders, Crohn's disease (CD) and ulcerative colitis (UC). The exact aetiology of IBD is not well known. There are several factors involved in development of this group of diseases, which include changes in the immune system, bacterial infection and genetic variations [1]. CD is characterized by discontinuous transmural inflammation that, although can occur in any region of gastrointestinal tract, strikes mostly the ileum and the colon. On the contrary, in UC the inflammatory state, typically limited to the mucosa, involves the rectum and may affect a part or the entire colon in a continuous pattern [2,3]. Table 1 shows the highest incidence and prevalence of IBD in principal areas of the world. It is evident that these parameters are more prevalent in highly industrialized nations than in less developed countries [4,5]. The lifestyle and dietary factors affect the development of IBD. In fact, the cigarette smoke enhances the risk for CD and influences negatively the life quality of patients [6,7]. The excessive consumption of milk, animal proteins, carbohydrates and polyunsaturated fatty acids (PUFAs) can enhance the risk for IBD. In particular, starch, generally present in human diet, can promote development of microbic species involved in the induction of autoimmunological reaction in IBD [8]. It has been observed that the imbalance in the consumption of Omega 6 PUFAs and Omega 3 PUFAs is very important in the development of IBD. In fact, Omega 3 PUFAs, differently from Omega 6 PUFAs, do not alter parameters, such as intestinal microbic flora and intestinal permeability, which contribute to IBD pathogenesis [9]. In the development of IBD, an important role is attributed to oxidative stress, due to altered balance between reactive oxygen species (ROS) and antioxidant activity [10]. In particular, the increased oxidative state in intestinal mucosa of CD patients is involved in the chronicization of this pathology [11,12]. Recently, it has been demonstrated that the compounds with antioxidant capacity, associated or not to the traditional therapy for IBD, such as sulfasalazine, azatriopine, corticosteroids, 5-aminosalicylic acid and infliximab, can be beneficial for IBD treatment [13,14]. The compounds used in the antioxidant therapy are polyphenols, such as curcumin, resveratrol, quercetin, cathechin, present in plants and in food originating from plants [15]. Also food rich in micronutrients with antioxidant properties, such a vitamin E and C, can ameliorate ROS-induced symptoms [16]. Proteinases, such as matrix metalloproteinases (MMPs), and A disintegrin and metalloproteinase 17 (ADAM17), belonging to ADAM metalloproteinases, affect the intestinal tissue damages and the inflammatory processes which characterize IBD [17-19]. In fact, the oxidative stress can play a crucial role in the modulation of MMPs activity and expression [20]. MMPs are involved principally in the turnover of extracellular matrix, while ADAM17, a membrane-bound enzyme, cleaves cell surface proteins such as cytokines and cytokine receptors [21]. Deregulated activity and expression of MMPs affect cell adhesion, immune cell migration, cytokine production and impairs wound healing [22]. The activities of MMPs are closely regulated at various levels such as transcription, activation of zymogens and inhibition by endogenous inhibitors named tissue inhibitors of MMPs (TIMPs) [23,24]. TIMPs reduce MMPs activity, by forming non covalent 1:1 stoichiometric complex, [25] but they also affect, through their specific receptor, cell growth, apoptosis, differentiation and angiogenesis [26]. The balance between MMPs and TIMPs is very important in the inflammatory response and in the outcome of tissue injury. In fact, an increased MMPs/TIMPs ratio has been found in inflamed colon of IBD [27], and an enhanced activity of MMPs may amplify intestinal immune response by recruiting the inflammatory cells [28]. Given that, it has been suggested the potential use of inhibitors of MMPs in IBD treatment as demonstrated in IBD animal models in which the administration of several MMP inhibitors reduces the inflammatory state [29,30]. Moreover, some data indicate that the antioxidant compounds may have a therapeutic effect by reducing the altered levels of MMPs detected in intestinal subepithelial myofibroblasts (ISEMFs) isolated from colonic mucosa of CD patients (CD-ISEMFs) [31,32]. An increase of ADAM17 has been also found in human colonic mucosa of IBD patients [33] and this suggests that ADAM17 inhibition may be an alternative strategy to block signalling pathway of tumor necrosis factor (TNF-α), a cytokine involved in intestinal inflammation.
![]()
Table 1: Highest annual percent of incidence and prevalence of ulcerative colitis (UC) and Crohn's disease (CD) in the principal areas of the world.
View Table 1
Metalloproteinases
MMPs, collectively called matrixins, participate in the modulation of homeostasis of extracellular matrix (ECM) by cleaving ECM proteins such as collagen, fibronectin, elastin and laminin [34]. However, even if MMPs are involved in the normal tissue remodelling, they also act on substrates different from ECM proteins, regulating many physiological functions such as cell proliferation, adhesion, migration, growth factor bioavailability, chemotaxis and signalling [34]. Under normal physiological conditions, MMPs are present in the latent form and at low concentration. They generally present a prodomain, a catalytic domain, a hinge region and a hemopexin domain [34]. To date 23 MMPs have been found and identified in humans. On the basis of substrate specificity, sequence similarity and domain organization, MMPs can be divided into: Collagenases, Gelatinases, Stromelysins, Matrilysins, Membrane-Type MMPs. These last MMPs distinguish in transmembrane or glycosylphosphatidylisositol- (GPI)-anchored MMPs (Table 2). Seven MMPs are not classified in the above categories and have been indicated as other MMPs. In particular, MMP-12 (metalloelastase), mainly expressed in macrophages, is essential for their migration and digests, besides elastin, a number of other proteins [35,36]. MMP-19 is identified as T-cell-derived autoantigen from patients with rheumatoid arthritis [37]. MMP-20, located within newly formed tooth enamel, is also called enamelysin and digests primarily amelogenin [38]. MMP-21 has been detected in neuronal tissues [39], in cancer and fetal tissue [40], indicating a role in human embryogenesis and tumor progression. MMP-22 function is still not known, but its substrates "in vitro" are gelatin and casein [41]. The characterization of MMP-21 and MPP-22 genes showed almost identical sequences [42]. MMP-23, mainly expressed in reproductive tissues, is a membrane-anchored protein with four domains. Differently to other MMPs, it lacks the cysteine switch motif in the prodomain and also the hemopexin domain. Instead, it has a cysteine-rich domain followed by an immunoglobulin-like domain [43]. The latest member added to MMP family is MMP-28, epilysin, initially found in keratinocytes. It is expressed in a number of normal tissues, suggesting that it regulates tissue homeostasis [44]. Enhanced expression of epilysin has been observed in basal keratinocytes during wound healing; in fact, expression patterns in intact and damaged skin suggest that MMP-28 might function both in tissue homeostasis and wound repair [45].
![]()
Table 2: Classification of different MMPs on substrate specificity.
View Table 2
Regulation of MMPs
Catalytic activity of MMPs is regulated at four levels: gene expression, compartmentalization, proenzyme activation, enzyme inactivation. Moreover, it is further controlled by substrate availability and affinity. Some MMP members, such as MMP-2, MMP-19, MMP-28 and several MT-MMPs, are expressed in normal tissue, being involved in tissue homeostasis. However, most MMPs are induced in tissue repair or remodelling subsequently to diseases or inflammatory processes. For the most part, the production of MMPs is regulated at transcription level by specific signals that are temporally limited and spatially confined [34]. MMPs are secreted as proenzymes and are activated in the pericellular and extracellular space. One-third of MMPs contains or RRKR sequence between the pro and catalytic domain, which serves as a target sequence for proprotein convertases (PPCs), a family of proteins that activate other proteins. One of the most well-known PPCs is Furin, a subtilisin-like serine protease present in trans-Golgi network [46], which cleaves protein precursors on motifs such as Arg-X-X-Arg and Lys/Arg-Arg. In fact, MMPs with a furin cleavage site are processed intracellularly before secretion [47,48].
In MMP prodomain is present a conserved cysteine that interacts by its thiol group with the zinc ion of the catalytic site. In this way, ProMMPs are kept in the inactive state and become catalytically active after disruption of the thiol-Zn2+ interaction [34]. This process, named the "cysteine-switch", represents a general and required step in the activation of all proMMPs [49]. The break of thiol-Zn2+ interaction can be due to the proteolytic activity of other proteinases that directly remove the prodomain. ROS, SH-reactive agents, or detergents can also induce conformational changes of the proenzyme, thus favouring conversion to an activated state by autolytic cleavage of the prodomain [50] (Figure 1). However, allosteric perturbation, due to binding of proMMPs to macromolecules, such as integrins, gelatin, collagen, can activate MMPs in their intact or partially cleaved form [51]. In fact, in the cysteine-switch mechanism, the removal of prodomain is not necessary for the activation of proMMPs; differently, the elimination of zinc-thiol interaction is required. Oxidants firstly activate, by oxidating the thiol prodomain, and subsequently inactivate MMPs, by modifying amino acids crucial for their catalytic activity. In particular, ROS, by modifying cysteine SH-group, activate proMMPs via autolytic cleavage "in vitro" [52]. However, this ROS effect occurs at low concentrations; in fact, high concentrations of ROS induce the inactivation of MMPs [53]. Also S-glutathionylation of the cysteine in the conserved domain of propeptide that occurs in the presence of peroxynitrite and glutathione (GSH), activates MMPs without cleavage of prodomain [54]. On the contrary, other studies show that MMPs activation, due to peroxynitrite alone or with GSH, is followed by the cutting of prodomain [55], indicating that both mechanisms are possible.
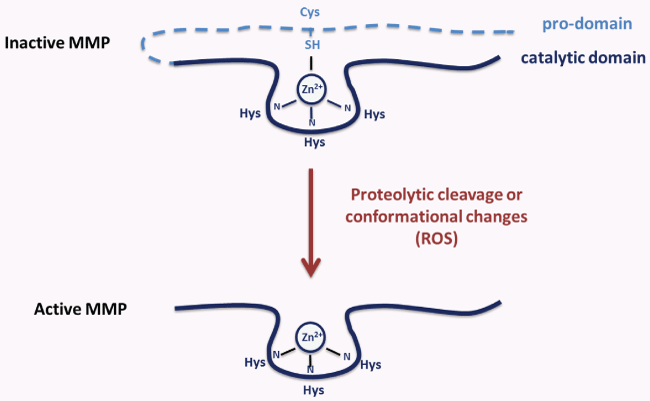
.
Figure 1: MMP activation: involvement of "cysteine-switch" mechanism.
MMPs are activated through pro-domain proteolytic cleavage or conformational changes. Reactive oxygen species (ROS) break thiol-Zn2+ interaction and induce conformational changes which favor MMP activation.
View Figure 1
The local tissue activities of MMPs are also modulated by TIMPs, specific inhibitors of matrixins [56,57]. Four TIMPs (TIMP-1, TIMP-2, TIMP-3 and TIMP-4) have been identified in vertebrates [58], and their expression is regulated during development and tissue remodelling. Under pathological conditions, changes in TIMP levels are very important considering that they directly affect and unbalance MMP activity. TIMPs have an N- and C- terminal domain of about 125 and 65 amino acids respectively, each containing three conserved disulfide bonds [59,60]. The N-terminal domain folds as a separate unit and is able to inhibit MMPs [59]. TIMP-1 structure and its inhibition mechanism were determined by X-ray crystallographic studies of the TIMP-1-MMP-3 complex [61]. TIMPs inhibit all MMPs tested so far, except for TIMP-1 that failed to inhibit MT1-MMP [62].
MMPs in IBD
Collagenases (MMP-1, MMP-13)
An increased expression of MMP-1, related to degree of inflammation, has been detected in biopsies from colonic mucosa of patients with active IBD [27,63]. In particular, MMP-1 expression increases by 230-fold in ulcerated colonic mucosa of IBD patients as compared to normal controls. Moreover, the increase of MMP-1 is attributed principally to macrophages present within the inflamed mucosa [27]. The enhanced expression of MMP-1 potentiates the inflammatory response in IBD intestinal ulcers [64] and in CD granulation tissue [65, 66], suggesting that MMP-1 plays an important role in the increase of inflammatory state. The increased expression of MMP-1 in the colonic mucosa of UC patients, related to the enhancement of MMP-1 plasma levels [67], induced acute tissue injury and initial steps of ulceration due to an excessive cleavage of ECM proteins [68]. Indeed, the expression of MMP-1 is greatly increased in ulcerated and inflamed colon areas and in serum of UC patients [67,69-71]. All together, these results suggest that MMP-1 expression and release correlate with UC severity and mucosal damage. Analysis of the relationship between MMP-1 expression and the severity of the disease shows that the expression of MMP-1 in patients with moderate UC is significantly higher than that in patients with mild UC, demonstrating that MMP-1 can be used as biomarker to judge the severity of clinical symptoms in patients. An enhanced expression of MMP-1 up-regulates TIMP-1 in ulcerated mucosa of UC patients, but this leads to an imbalance between MMP-1 mRNA and TIMP1 mRNA levels. In reality, TIMP-1 mRNA levels do not counteract those of MMP-1 mRNA with consequent overdegradation of ECM in UC [68].
MMP-13 is another collagenase that has as substrate also pro-MMP-9, and is considered important in IBD being strongly expressed in fibrotic areas of chronic cutaneous ulcers in IBD patients. In particular, it is detected only in fibroblasts of the ulcer bed, for this it could be involved in remodelling of the submucosal matrix [72]. The pro-inflammatory role of MMP-13 is due to its ability to release TNF-α from membranes [73]. In fact, it has been demonstrated that both the administration of dextran sulphate sodium (DSS) in rats, in order to mimic colitis clinical conditions, and lipopolysaccharide (LPS)-induced sepsis up-regulate MMP-13 levels involved in the TNF-α cleavage [74]. The formation of bioactive TNF-α causes alterations of intestinal epithelial barrier integrity by increasing intestinal permeability and destabilizing tight junctions [74]. To our knowledge there are no data about the role of oxidative stress on collagenase activity in IBD. However, some data in literature show that an increase of MMP-1 expression and activity, related to ROS production, occur in a variety of cell lines and tissues [75]. Also buthionine sulfoximine (BSO), inhibitor of GSH synthesis, and aminotriazol, catalase inhibitor, increase MMP-1 expression [76]. MMP-13 is overexpressed in osteoarthritic chondrocytes when an increase of ROS production occurs [77]. On the contrary, antioxidants such as the polyphenol epigallocatechin-3-gallate, present in green tea, GSH, Vitamin A and E decrease MMP-1 expression in liver fibrosis, in transformed human heart fibroblasts and in a porcine model of atherosclerosis [78-80]. Moreover, resveratrol, polyphenolic compound present in red wine with anti-inflammatory and antioxidant properties [81], inhibits production of MMP-1 and stromelysin MMP-3 in human chondrocytes [82], and MMP-13 expression in osteoarthritic rats [83].
Gelatinases (MMP-2, MMP-9)
MMP-2 is expressed in normal colon, mainly in the epithelial cells and in lamina propria. An increase of MMP-2 activity was detected in IBD [17,63] and in particular in CD intestinal fistulae [84]. In fact, the increased proteolytic activity of MMP-2 can have an important role in fistulae formation by degrading the basement membrane that supports endothelial and epithelial cells in the gut [84]. The epithelial barrier dysfunction characterizes the pathogenesis of intestinal inflammation and MMP-2 represents a key factor for intestinal barrier functionality [85,86]. However, the role of MMP-2 in the development of colitis is still unclear. Some authors demonstrated that MMP-2 plays an important protective role in maintenance of the integrity of intestinal epithelial barrier [87]. They show that in experimental animal models of colitis, induced by chemicals or bacteria, MMP-2-/- mice develop the acute colitis much more intensely than WT mice [87]. Myofibroblasts are an important source of MMP-2 in intestine during the inflammatory state [84]. An increase of MMP-2 secretion and activation, related to decrease glutathione/oxidized glutathione ratio (GSH/GSSG) and inflammatory state, is detected in CD-ISEMFs [31]. In these cells, N-acetylcysteine (NAC), a precursor of GSH synthesis, down-regulates MMP-2 secretion through the increase of GSH/GSSG ratio and exhibits a direct inhibitory action on MMP-2 activity. Moreover, NAC is also able to reduce MMP-2 activation induced by TNF-α thus restoring the physiological activity of enzyme in CD-ISEMFs [31]. Redox regulation of MMP-2 secretion involves c-jun N-terminal kinase (JNK) pathway [31] that phosphorylates transcriptional factors present in most MMP promoters [88]. All data on MMP-2 indicate the importance of correct regulation of production and activity of this MMP. In fact, the lack of MMP-2 can be responsible for deregulation of intestinal barrier function; on the contrary, the excess of MMP-2 can induce fistulae formation. The role of oxidative stress on secretion and activation of MMP-2 has been confirmed in CCD-18Co cells (18Co), a myofibroblast cell line derived from human colonic mucosa, in which the intracellular redox state was modulated through BSO or NAC [31].
High levels of MMP-9 are also present in several animal models of colitis and in human IBD [63,89]. Experimental DSS-induced colitis in rats increases pro-MMP-9 activity [29]. The synthetic inhibitor of MMPs, CGS-27023, administrated to these rats for 14 days, increases mucosal repair process and reduces inflammation, indicating a close relationship between pro-MMP-9 activity and injury in inflamed colonic tissue [29]. MMP-9 is the most abundantly expressed protease that correlates with disease activity in IBD [17,90], and it is associated with fistulae in acute CD [17,91]. It has been demonstrated, by immunohistochemical analysis, that MMP-9 is expressed abundantly in fistulae of patients with CD, but only in those with prominent acute inflammation [84]. In the fistulae formation, MMP-9 could have a similar role to that of MMP-2; however, MMP-9 may promote tissue injury by inducing extravasation of neutrophils in areas with acute inflammation and by accelerating the proteolysis of matrix proteins partly degraded by other MMPs [91]. These tissue damages play a pivotal role in IBD; in fact, differently to that occurs for MMP-2, MMP-9-/- mice do not develop colitis due to bacteria or chemicals [86] and are resistant to inflammatory processes [29,92,93]. MMP-9 is involved in the inflammatory response by delaying re-epithelialization processes [94], impairing wound healing [95], increasing endothelial permeability [96,97], and activating several proteins including fibrinogen, interleukin (IL)-1β, IL-8, and transforming growth factor-β (TGF-β) [95,98-100]. It is shown that serum concentration of MMP-9 is higher in UC and CD patients than that measured in control serum, and MMP-9 levels correlate well with the disease activity [101]. These data were confirmed by the demonstration that a significant increase of MMP-9 occurs in active CD and UC as compared with MMP-9 levels detected in controls and in patients with inactive forms [102]. The correlation between MMP-9 serum levels and the clinical activity of disease was observed also in children affected by IBD [103,104]. Fecal MMP-9 levels increase in IBD patients but this increase is higher in UC than in CD [105]. These results suggest that serum and fecal MMP-9 levels can be considered as supportive markers for disease activity or for diagnosis of different types of IBD, respectively. An up-regulation of MMP-9 expression has been associated with the release of vascular endothelial growth factor (VEGF) in animal models of colitis [106]. In fact, a strong correlation between circulating VEGF-A and serum MMP-9 in CD patients was observed [102]. MMP-9 seems to be involved in angiogenesis by recruitment of pericytes to the new vessels [107]. In this process platelet-derived growth factor (PDGF), whose availability as well as biological functions are regulated by its binding to ECM, plays the major role. Given that a correlation between the concentration of this growth factor and MMP-9 levels in both CD and UC was found, it has been speculated that MMP-9 might participate in the angiogenesis through the release of PDGF from ECM or degradation of ECM elements involved in PDGF binding [102]. Recent data demonstrate that aerial part extracts of Lavandula dentata and Lavandula stoechas improve the redox state by increasing GSH levels in 2,4,6-trinitrobenzene-sulfonic-acid (TNBS)-induced rat colitis model [108]. Moreover, this increase of redox state is associated to down-regulation of MMP-9 expression [108].
Stromelysins (MMP-3, MMP-10)
MMP-3 is induced consistently in several animal models [109] and in human IBD [63]. It is expressed during IBD in mononuclear macrophage-like cells and in lamina propria stromal cells below the ulcerated areas [38,72,110]. An up-regulation of MMP-3 is present in CD [65,110] and, similarly to gelatinase MMP-9, it is related to mucosal damage and fistulae formation through degradation of the matrix in areas with active inflammation [17,73,91]. MMP-3 and MMP-9 concentrations in sera of pediatric patients with IBD reflect various stages of disease activity. In fact, significantly higher concentrations of these MMPs are present in the sera of patients with the active form of disease as compared to those found in sera of patients with the mild form or in control group [103,104]. Therefore, the levels of MMP-3 and MMP-9 might be useful in the clinical evaluation of patients. A marked overexpression of MMP-3 was assessed by mRNA levels in inflamed mucosal samples from patients with UC as compared to controls [27]. These results are also supported by data that demonstrate an overexpression of MMP-3 in intestinal inflamed sites of children with IBD [111]. Functional experiments in an ex vivo human foetal small intestine explant model have clearly shown that MMP-3 also acts as a key mediator of T cells, and TNF-α mediates tissue injury in the gut [112-114]. The increase of oxidative state present in CD-ISEMFs is responsible for the increased MMP-3 production detected in these cells stimulated or not with TNF-α, as compared to the control ISEMFs [32]. In fact, NAC restores MMP-3 levels in CD-ISEMFs to those of control ISEMFs. Data obtained in 18Co treated with BSO and/or NAC or curcumin, antioxidant present in Curcuma Longa with anti-inflammatory effect [115], highlight the regulatory role of antioxidants on production of MMP-3. However, their effect is not closely related to changes of oxidative state, suggesting that antioxidants affect MMP-3 production also by their direct action on transcriptional factors [32]. Indeed, mitogen activated protein kinases (MAPKs), activated in the presence of oxidative stress, are involved in the modulation of MMP-3 levels [31,32]. Curcumin decreases MMP-3 expression both by increasing GSH levels and by inhibition of p38 mitogen activated protein kinase (p38 MAPK) signalling pathway and transcriptional factors, such as nuclear factor-kB (NF-kB) and activator protein-1 (AP-1) [116]. Moreover, curcumin reduces MMP-3 in biopsies and ISEMFs of adult and children affected by IBD [117] indicating the role of oxidative stress in the increased activity of MMPs in these patients.
MMP-10 together with matrilysin MMP-7 plays a role in intestinal wound healing [118], and MMP-10 is actively expressed in epithelial cells at the ulcer margin. This location and the finding that the same cells produce laminin-5 suggest an important role for MMP-10 in epithelial cell migration [72]. In addition, MMP-10 is also expressed in macrophage and lymphocyte-like cells in areas of inflammation but not in T lymphocytes. In fact, T lymphocytes do not express MMP-10 in the intestine but stimulate other cells of lamina propria to express it, participating in formation of early lesions in IBD [72,112].
Matrilysin (MMP-7, MMP-26)
MMP-7, expressed in many adult tissues, is implicated in innate intestinal defence [119]. Cytokines and growth factors, such as IL-1, epidermal growth factor (EGF) and TNF-α, at elevated concentrations at inflammatory sites, may induce the synthesis of MMP-7 [120]. The expression of this MMP is an important hallmark of surface epithelium at the edge of gastrointestinal ulcers. In fact, MMP-7 was not detected in intact tissue distant from the wound [65] and in normal gastric or colonic mucosa [121,122]. MMP-7 mRNA was detected in crypt abscesses associated with idiopathic IBD [65]. Its expression also increases in IBD and its levels correlate with disease activity in UC [123,124]. During IBD, MMP-7 has been detected in areas surrounding ulcers, suggesting a role in repair/wound healing. Recently, it has been demonstrated that the glandular epithelium MMP-7 correlates with the disease incidence in UC and with the lesion location in CD [125].
MMP-26, specifically expressed in epithelial carcinomas and in normal adult kidney, decreases after anti-TNF-α treatments in intestinal neutrophils of CD patients [126]. This suggests the involvement of MMP-26 in tissue destruction or migration of neutrophils. MMP-26 is also expressed in migrating enterocytes localized next to the intestinal ulcer in IBD [127].
Other MMPs (MMP-12, MMP-19, MMP-28)
MMP-12 increases in the immune response in lamina propria macrophages [72,128], and this proteinase may have a role in macrophage migration and tissue inflammation in CD [128]. MMP-19, expressed in non-migrating enterocytes in IBD intestine [127], seems to be important for appropriate immune response in colitis. In fact, in DSS-induced colitis the lack of MMP-19 causes an increase of inflammatory state and mucosal damage in mice [129]. MMP-28, differently to previously described MMPs, decreases in inflamed mucosa in UC [130], suggesting that this MMP is not involved in IBD intestinal damage [127].
ADAM17 and its regulation
The differences between MMPs and ADAMs consist in domain structure and in particular in the sequence of catalytic domain. The ADAM structure contains a prodomain, a metalloproteinase catalytic domain, a disintegrin domain, an EGF-like (cysteine-rich) domain, a single transmembrane domain and a cytoplasmic portion [21] (Figure 2). Members of ADAM family have emerged as major ectodomain shedding proteinases, which release a large number of molecules in various physiological conditions. However, the uncontrolled release of some substrates is related to various pathological processes. ADAM17, also known as tumor necrosis factor-α converting enzyme (TACE), is a membrane-bound enzyme belonging to ADAM family that cleaves cell surface proteins such as cytokines (TNF-α) or cytokine receptors (IL-6-receptor and TNF-receptor). Considering the effect of ADAM17 on TNF-α or IL-6, it has been speculated that its inhibition could improve autoimmune diseases [131]. In fact, membrane-bound TNF-α acquires the pro-inflammatory activity after its cleavage by ADAM17 [132]. Moreover, the shedding of IL-6R from membrane induces IL-6 trans-signalling on cells expressing only IL-6 receptor chain gp130, thus increasing the inflammatory state with consequent cancer risk [133,134]. Phorbol ester, phorbol 12-myristate 13-acetate (PMA), is a potent activator of ADAM17 by protein kinase C (PKC) and extracellular signal-regulated kinase (ERK) activation. In fact, the inhibition of these kinases decreases ADAM17 activity [135]. ROS induce an increase of ADAM17 activity through direct modification of disulphide bonds that allows ADAM17 to assume an open active conformation [136] and also mediate up-regulation of ADAM17 expression [137,138]. The compartmentalization plays an important role in the regulation of ADAM17. In fact, it is stored in perinuclear region and its translocation on cell membrane occurs after phosphorylation of Thr 735 modulated by p38 MAPK activation [139]. Apoptosis also induces, probably by activation of p38 MAPK, an increase of ADAM17 activity and consequent translocation on cell surface [140]. ADAM17 activity is down-regulated by the natural tissue inhibitor TIMP-3 [141], and it has been demonstrated that also Tetraspanin CD9 negatively regulates ADAM17-mediated TNF-α shedding as well as intercellular adhesion molecule-1 on the cell surface [142].
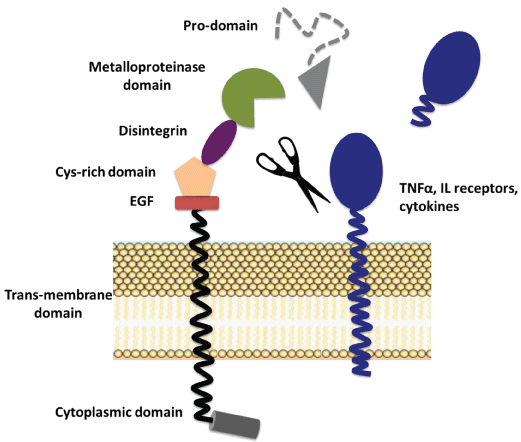
.
Figure 2: Schematic structure and activation of ADAM members.
Members of ADAM family after activation cleave the substrates from the cell surface.
View Figure 2
Members of adam family in IBD (ADAM9, ADAM10, ADAM17, ADAM19)
ADAM17, normally expressed in the human colonic mucosa, increases in human IBD [33] and its expression is up-regulated by TNF-α in endothelial cells [143]. Transmigration of polymorphomononuclear leukocytes (PMNL) across epithelia is the primary event of acute phase in CD, and it can be responsible for the TNF-α secretion by intestinal cells [144]. Moreover, in this phase of acute inflammation, an increase of the expression of ADAM17, but not of its inhibitor TIMP-3, occurs in intestinal endothelial cells (IEC) [145]. All these data demonstrate that the early upregulation of ADAM17 was linked to TNF-α production during PMNL transepithelial migration. Reduced levels of ADAM17 increase sensitivity to colitis in mice [146] and studies, performed in DSS-induced colitis, demonstrate that epithelial ADAM17 is very important to provide colitis resistance in rats [147]. In fact, it is possible that ADAM17, through the activation of EGFR and its signalling pathways, promotes mucosa repair by inducing epithelial cells proliferation and globet cell differentiation [147]. Some data show that the ADAM17 expression levels, detected in epithelial cells and in intestinal biopsies derived from intestinal mucosa of IBD patients, were not significantly different from ADAM17 values measured in control samples [148,149]. On the contrary, other data demonstrate that ADAM17 expression enhances in CD patients and promotes neutrophil migration and colitis [145]. It is possible that the differences highlighted in these studies may be due to different inflammatory state of intestinal mucosa and/or therapy used. In fact, ADAM17 levels can be related to oxidative stress considering that an increase of this protease occurs in intestinal myofibroblast cell line in which is present an increase of ROS due to BSO and/or TNF-α treatment [138]. The involvement of ADAM17 in IBD is also observed in mice with DSS-induced colitis, in which TACE significantly increases during the period of DSS treatment and returns to normal levels in the remission phase. Moreover, the treatment with a specific inhibitor of ADAM17, compound 11p, reduces clinical signs of DSS-induced colitis maintaining the integrity of colon mucosa and avoiding the infiltration of inflammatory cells [150]. Resveratrol ameliorates experimental colitis by reducing ADAM17 and increasing TIMP-3 expression through the activation of the deacetylase, silent information regulator-1 (SIRT-1) [150]. ADAM17 can be considered a target in therapies for chronic inflammatory diseases and compounds able to inhibit ADAM17 activity have already been developed [151].
Although ADAM17 is the member of ADAM family highly expressed in IBD, it has been demonstrated that ADAM19 is also up-regulated in mucosa of IBD patients [152]. The major increase of ADAM19 is detected in patients with UC and its expression is stimulated by TNF-α and various cytokines. Differently, no variation of ADAM9 and ADAM10 expression between IBD patients and control was observed [152].
TIMPs in IBD (TIMP-1, TIMP-2, TIMP-3, TIMP-4)
TIMP-1: Contradictory data are reported in literature about the production of TIMP-1 in intestinal inflamed mucosa. In IBD it has been demonstrated that TIMP-1, expressed by inflammatory cells, activated fibroblasts and vascular smooth muscle cells, increases predominantly in the areas of mucosa with active inflammation and, in particular, at ulcer bases [64]. This localization suggests that TIMP-1, along with the increase of MMP-1, plays a role in the tissue reorganization after mucosal damage. An increased expression of TIMP-1, related to disease severity, is observed in inflamed intestinal area of UC patients [69], as well as in inflamed and especially ulcerated colon mucosa of IBD patients [27]. In a study using cultures of biopsies isolated from intestinal mucosa of IBD patients, TIMP-1 levels were higher in inflamed mucosa of both CD and UC as compared with uninflamed mucosa [153]. Moreover, TIMP-1 showed a strong correlation with levels of proinflammatory cytokines such as IL-6, IL-1β and IL-10 [153]. An enhancement of TIMP-1 has been observed both in inflamed intestinal mucosa and in serum of UC patients [71]. In particular, it has been observed that TIMP-1 plasma levels in UC patients correlate positively with scored endoscopic degree of mucosal injury, with indices of disease and clinical activity, and with C reactive protein concentration, a marker of inflammation levels. Therefore, TIMP-1 plasma concentration may be a possible biomarker of disease activity [71]. No increase of TIMP-1 production has been detected in CD-ISEMFs type cells stimulated or not with TNF-α as compared to C-ISEMFs [32]. Similar results were obtained in 18Co treated with BSO and/or NAC indicating that the intracellular redox state does not affect the production of this inhibitor [32]. Indeed, an increased secretion of TIMP-1 is measured only in fibrotic myofibroblasts and is associated with the presence of fibrotic structures in CD [154]. Moreover, it has also been observed that TIMP-1 production enhances in myofibroblasts of CD patients only after treatment with infliximab (chimeric monoclonal antihuman TNF-α antibody), and this effect is not accompanied by significant induction of apoptotic process or alterations in MMP expression [155]. Considering these findings, it has been hypothesized that not myofibroblasts but other inflammatory cells, such as lymphocytes and macrophages, may be the responsible source for the increased levels of TIMP-1 in intestinal mucosa of CD patients [27,32].
TIMP-2: No statistical difference in TIMP-2 levels was found between IBD patients and healthy controls in serum [156]. Moreover, no remarkable expression of TIMP-2 is observed in inflamed mucosa of IBD patients [156]. TIMP-2 mRNA levels, measured by PCR in biopsies from IBD patients, remain unchanged in inflamed and uninflamed mucosa [27]. On the contrary, other studies demonstrated an increase of TIMP-2 serum levels in UC and CD patients, suggesting its use as a potential marker for IBD activity [157].
TIMP-3: TIMP-3, present in both uninflamed and inflamed gut mucosa and predominantly expressed in macrophage-like cells or fibroblastic-like cells in lamina propria and in endothelial cells [72], inhibits the inflammatory response as shown by studies based on timp3-/- mice or gene transfer "in vitro". TIMP-3 is essential for innate immune function and has a pivotal role in the regulation of inflammatory response [158]. In fact, the loss of this TIMP causes deregulation of cleavage of TNF-α and its receptors by inactivation of ADAM17 in innate immunity [158]. Very severe colitis, associated with increased expression of inflammatory cytokines, has been determined in TIMP-3-deficient mice after treatment with TNBS [149]. In contrast, transgenic mice for TIMP-3 express less cytokines and are largely resistant against TNBS-induced colitis. In addition, it has been demonstrated that TIMP-3 regulates negatively not only ADAM17 but also functional activity of MMPs that are increased in IBD [149]. For this reason, given that MMP-9 derived from epithelium has a crucial role in the induction of intestinal damage, the severe intestinal damage seen in phenotype TIMP-3-KO mice after TNBS administration, may be due to the lack of inhibitory effect of TIMP-3 on MMP-9. However, there are no commercial compounds that can selectively inhibit MMP-9 and ADAMs in order to assess the exact contribution of these two different proteases in colitis occurring in TIMP-3-KO mice. A decreased expression of TIMP-3 has been detected in DSS-induced colitis in rats [159] and in inflamed intestine of CD patients [149]. These data are in contrast with the up-regulation of TIMP-3 that occurs in condition of oxidative stress in macrophages and in chondrocytes [160,161]. In particular, the up-regulation of TIMP-3 expression, related to ROS production and suppressed by NAC treatment is mediated by small mother against decapentaplegic (Smad) 2 protein [161]. Considering that TGF-β1 increases TIMP-3 expression also in normal gut, it is possible that the diminished TIMP-3 expression in CD patients is in part due to Smad7, an inhibitor of TGF-β1 signalling, expressed at high levels in CD mucosa [149]. TIMP-3 decrease can be responsible of the increased paracellular permeability and tight junction protein alteration due to TNF-α [148]. Taken all, these findings suggest that TIMP-3-based therapies capable of blocking TNF-α may be considered in clinical use to control IBD in TGF-β1-stimulated chondrocytes
TIMP-4: It is shown that the levels of TIMP-4 decrease in serum of IBD patients with a consequent increase of MMP-2 activity. This may be very important, because MMP-2 is a key player in proper wound healing, angiogenesis and re-epithelization, as well as in the regulation of epithelial barrier function of the intestine [156]. TIMP-4 also inhibits platelet aggregation suggesting its involvement in the regulation of platelet recruitment and aggregation, processes enhanced in IBD patients and even in inactive disease [162,163]. Table 3 summarizes the principal roles of MMPs, ADAM17 and TIMPs in IBD.
![]()
Table 3: MMPs, ADAMs and TIMPs expression and role in inflammatory bowel disease (IBD).
View Table 3
Conclusions
MMPs, ADAMs and TIMPs play an important role in wound healing and in numerous pathophysiological processes. Moreover, they are involved in the modulation of inflammatory response, by controlling migration of immune cells, matrix deposition and degradation as well as cytokine activity. However, an increase of MMPs and ADAM17, often linked to the disease severity and not accompanied by an up-regulation of TIMP, is present in intestinal mucosa of IBD patients. This can contribute to increase the inflammatory state, cytokine production, ulceration and fistulae formation. MMPs, differently to TIMPs, are also redox-regulated. This review highlights that the up-regulation of some MMPs production in IBD can be due to the oxidative stress present in intestinal mucosa of IBD patients. In fact, antioxidants are able to down-regulate MMPs production restoring their levels to those of control. Considering that an important role is attributed to oxidative stress in the pathogenesis of IBD, it is possible that the imbalance between oxidants and antioxidants can affect the expression of various MMPs in IBD. Moreover, antioxidants, in addition to restoring the intracellular redox state, act directly on the regulation of MAPKs and transcriptional factors (Figure 3). For this, in the development of MMP-targeting therapies, molecules with antioxidant property may be used to reduce dysfunction of epithelial barrier and prevent fistulae and ulceration in IBD.
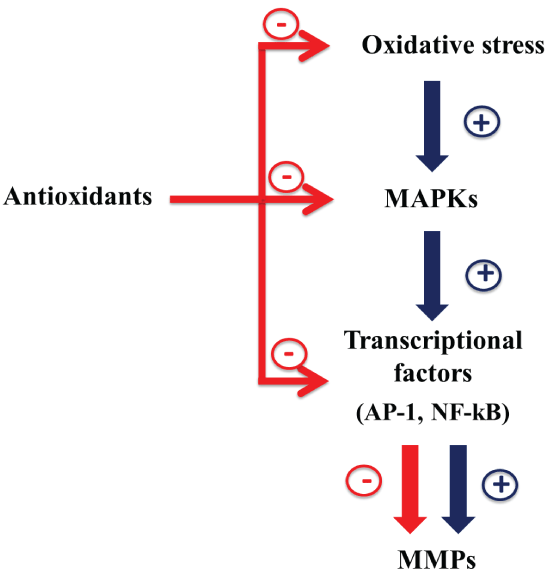
.
Figure 3: Down-regulation of MMPs expression by antioxidants: possible mechanisms.
View Figure 3
Conflict of Interest
The authors have no conflict of interest.
References
-
Abraham C, Cho JH (2009) Inflammatory bowel disease. N Engl J Med 361: 2066-2078.
-
Fakhoury M, Negrulj R, Mooranian A, Al-Salami H (2014) Inflammatory bowel disease: clinical aspects and treatments. J Inflamm Res 7: 113-120.
-
Corridoni D, Arseneau KO, Cominelli F (2014) Inflammatory bowel disease. Immunol Lett 161: 231-235.
-
Abdul Rani R, Raja Ali RA, Lee YY (2016) Irritable bowel syndrome and inflammatory bowel disease overlap syndrome: pieces of the puzzle are falling into place. Intest Res 14: 297-304.
-
Molodecky NA, Soon IS, Rabi DM, Ghali WA, Ferris M, et al. (2012) Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 142: 46-54.
-
Hovde O, Moum BA (2012) Epidemiology and clinical course of Crohn's disease: results from observational studies. World J Gastroenterol 18: 1723-1731.
-
Quezada SM, Langenberg P, Cross RK (2016) Cigarette smoking adversely affects disease activity and disease-specific quality of life in patients with Crohn's disease at a tertiary referral center. Clin Exp Gastroenterol 9: 307-310.
-
Wedrychowicz A, Zajac A, Tomasik P (2016) Advances in nutritional therapy in inflammatory bowel diseases: Review. World J Gastroenterol 22: 1045-1066.
-
Uranga JA, Lopez-Miranda V, Lombo F, Abalo R (2016) Food, nutrients and nutraceuticals affecting the course of inflammatory bowel disease. Pharmacol Rep 68: 816-826.
-
Balmus IM, Ciobica A, Trifan A, Stanciu C (2016) The implications of oxidative stress and antioxidant therapies in Inflammatory Bowel Disease: Clinical aspects and animal models. Saudi J Gastroenterol 22: 3-17.
-
Iantomasi T, Marraccini P, Favilli F, Vincenzini MT, Ferretti P, et al. (1994) Glutathione metabolism in Crohn's disease. Biochem Med Metab Biol 53: 87-91.
-
Pinto MA, Lopes MS, Bastos ST, Reigada CL, Dantas RF, et al. (2013) Does active Crohn's disease have decreased intestinal antioxidant capacity? J Crohns Colitis 7: e358-366.
-
Sandborn WJ, Feagan BG, Lichtenstein GR (2007) Medical management of mild to moderate Crohn's disease: evidence-based treatment algorithms for induction and maintenance of remission. Aliment Pharmaco. Ther 26: 987-1003.
-
Kannan N, Guruvayoorappan C (2013) Protective effect of Bauhinia tomentosa on acetic acid induced ulcerative colitis by regulating antioxidant and inflammatory mediators. Int Immunopharmacol 16: 57-66.
-
Moura FA, de Andrade KQ, dos Santos JC, Araujo OR, Goulart MO (2015) Antioxidant therapy for treatment of inflammatory bowel disease: Does it work? Redox Biol 6: 617-639.
-
Seifried HE, Anderson DE, Fisher EI, Milner JA (2007) A review of the interaction among dietary antioxidants and reactive oxygen species. J Nutr Biochem 18: 567-579.
-
Baugh MD, Perry MJ, Hollander AP, Davies DR, Cross SS, et al. (1999) Matrix metalloproteinase levels are elevated in inflammatory bowel disease. Gastroenterology 117: 814-822.
-
Gao Q, Meijer MJ, Kubben FJ, Sier CF, Kruidenier L, et al. (2005) Expression of matrix metalloproteinases-2 and -9 in intestinal tissue of patients with inflammatory bowel diseases. Dig Liver Dis 37: 584-592.
-
Arribas J, Esselens C (2009) ADAM17 as a therapeutic target in multiple diseases. Curr Pharm Des 15: 2319-2335.
-
Alge-Priglinger CS, Kreutzer T, Obholzer K, Wolf A, Mempel M, et al. (2009) Oxidative stress-mediated induction of MMP-1 and MMP-3 in human RPE cells. Invest Ophthalmol Vis Sci 50: 5495-5503.
-
Scheller J, Chalaris A, Garbers C, Rose-John S (2011) ADAM17: a molecular switch to control inflammation and tissue regeneration. Trends Immunol 32: 380-387.
-
Parks WC, Wilson CL, L0pez-Boado YS (2004) Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat Rev Immunol 4: 617-629.
-
Nagase H, Woessner JF Jr (1999) Matrix metalloproteinases. J Biol Chem 274: 21491-21494.
-
Sternlicht MD, Werb Z (2001) How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol 17: 463-516.
-
Gomez DE, Alonso DF, Yoshiji H, Thorgeirsson UP (1997) Tissue inhibitors of metalloproteinases: structure, regulation and biological functions. Eur J Cell Biol 74: 111-122.
-
Brew K, Nagase H (2010) The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. Biochim Biophys Acta 1803: 55-71.
-
von Lampe B, Barthel B, Coupland SE, Riecken EO, Rosewicz S (2000) Differential expression of matrix metalloproteinases and their tissue inhibitors in colon mucosa of patients with inflammatory bowel disease. Gut 47: 63-73.
-
Deem TL, Cook-Mills JM (2004) Vascular cell adhesion molecule 1 (VCAM-1) activation of endothelial cell matrix metalloproteinases: role of reactive oxygen species. Blood 104: 2385-2393.
-
Medina C, Videla S, Radomski A, Radomski MW, Antolin M, et al. (2003) Increased activity and expression of matrix metalloproteinase-9 in a rat model of distal colitis. Am J Physiol Gastrointest Liver Physiol 284: G116-G122.
-
Yamada A, Uegaki A, Nakamura T, Ogawa K (2000) ONO-4817, an orally active matrix metalloproteinase inhibitor, prevents lipopolysaccharide-induced proteoglycan release from the joint cartilage in guinea pigs. Inflamm Res 49: 144-146.
-
Romagnoli C, Marcucci T, Picariello L, Tonelli F, Vincenzini MT, et al. (2013) Role of N-acetylcysteine and GSH redox system on total and active MMP-2 in intestinal myofibroblasts of Crohn's disease patients. Int J Colorectal Dis 28: 915-924.
-
Fontani F, Marcucci T, Picariello L, Tonelli F, Vincenzini MT, et al. (2014) Redox regulation of MMP-3/TIMP-1 ratio in intestinal myofibroblasts: effect of N-acetylcysteine and curcumin. Exp Cell Res 323: 77-86.
-
Brynskov J, Foegh P, Pedersen G, Ellervik C, Kirkegaard T, et al. (2002) Tumour necrosis factor alpha converting enzyme (TACE) activity in the colonic mucosa of patients with inflammatory bowel disease. Gut 51: 37-43.
-
Ra HJ, Parks WC (2007) Control of matrix metalloproteinase catalytic activity. Matrix Biol 26: 587-596.
-
Shapiro SD, Kobayashi DK, Ley TJ (1993) Cloning and characterization of a unique elastolytic metalloproteinase produced by human alveolar macrophages. J Biol Chem 268: 23824-23829.
-
Shipley JM, Wesselschmidt RL, Kobayashi DK, Ley TJ, Shapiro SD (1996) Metalloelastase is required for macrophage-mediated proteolysis and matrix invasion in mice. Proc Natl Acad Sci U S A 93: 3942-3946.
-
Kolb C, Mauch S, Peter HH, Krawinkel U, Sedlacek R (1997) The matrix metalloproteinase RASI-1 is expressed in synovial blood vessels of a rheumatoid arthritis patient. Immunol Lett 57: 83-88.
-
Li W, Gibson CW, Abrams WR, Andrews DW, DenBesten PK (2001) Reduced hydrolysis of amelogenin may result in X-linked amelogenesis imperfecta. Matrix Biol 19: 755-760.
-
Marchenko GN, Marchenko ND, Strongin AY (2003) The structure and regulation of the human and mouse matrix metalloproteinase-21 gene and protein. Biochem J 372: 503-515.
-
Ahokas K, Lohi J, Lohi H, Elomaa O, Karjalainen-Lindsberg ML, et al. (2002) Matrix metalloproteinase-21, the human orthologue for XMMP, is expressed during fetal development and in cancer. Gene 301: 31-41.
-
Yang M, Kurkinen M (1998) Cloning and characterization of a novel matrix metalloproteinase (MMP), CMMP, from chicken embryo fibroblasts. CMMP, Xenopus XMMP, and human MMP-19 have a conserved unique cysteine in the catalytic domain. J Biol Chem 273: 17893-17900.
-
Gururajan R, Grenet J, Lahti JM, Kidd VJ (1998) Isolation and characterization of two novel metalloproteinase genes linked to the Cdc2L locus on human chromosome 1p36.3. Genomics 52: 101-106.
-
Galea CA, Nguyen HM, George Chandy K, Smith BJ, Norton RS (2014) Domain structure and function of matrix metalloprotease 23 (MMP23): role in potassium channel trafficking. Cell Mol Life Sci 71: 1191-1210.
-
Illman SA, Lohi J, Keski-Oja J (2008) Epilysin (MMP-28)--structure, expression and potential functions. Exp Dermatol 17: 897-907.
-
Saarialho-Kere U, Kerkela E, Jahkola T, Suomela S, Keski-Oja J, et al. (2002) Epilysin (MMP-28) expression is associated with cell proliferation during epithelial repair. J Invest Dermatol 119: 14-21.
-
Thomas G (2002) Furin at the cutting edge: from protein traffic to embryogenesis and disease. Nat Rev Mol Cell Biol 3: 753-766.
-
Illman SA, Keski-Oja J, Parks WC, Lohi J (2003) Mouse epilysin (MMP-28) is alternatively spliced and processed by a furin-like pro-protein convertase. Biochemistry 375: 191-197.
-
Kang T, Nagase H, Pei D (2002) Activation of membrane -type matrix metalloproteinase 3 zymogen by the proprotein convertase furin in the trans-golgi network. Cancer Res 62: 675-681.
-
Van Wart HE, Birkedal-Hansen H (1990) The cysteine switch: A principle of regulation of metalloproteinase activity with potential applicability to the entire matrix metalloproteinase gene family. Proc Natl Acad Sci USA 87: 5578-5582.
-
Springman EB, Angleton EL, Birkedal-Hansen H, Van Wart HE (1990) Multiple modes of activation of latent human fibroblast collagenase: Evidence for the role of a Cys 73 active-site zinc complex in latency and a "cysteine switch" mechanism for activation. Proc Natl Acad Sci USA. 87: 364-368.
-
Freise C, Erben U, Muche M, Farndale R, Zeitz M, et al. (2009) The alpha 2 chain of collagen type VI sequesters latent proforms of matrix-metalloproteinases and modulates their activation and activity. Matrix Biol 28: 480-489.
-
Fu X, Kassim SY, Parks WC, Heinecke JW (2001) Hypochlorous acid oxygenates the cysteine switch domain of pro-matrilysin (MMP-7). A mechanism for matrix metalloproteinase activation and atherosclerotic plaque rupture by myeloperoxidase. J Biol Chem 276: 41279-41287.
-
Fu X, Kao JL, Bergt C, Kassim SY, Huq NP, et al. (2004) Oxidative cross-linking of tryptophan to glycine restrains matrix metalloproteinase activity: specific structural motifs control protein oxidation. J Biol Chem 279: 6209-6012.
-
Okamoto T, Akaike T, Sawa T, Miyamoto Y, van der Vliet A, et al. (2001) Activation of matrix metalloproteinases by peroxynitrite-induced protein S-glutathiolation via disulfide S-oxide formation. J Biol Chem 276: 29596-29602.
-
Migita K, Maeda Y, Abiru S, Komori A, Yokoyama T, et al. (2005) Peroxynitrite-mediated matrix metalloproteinase-2 activation in human hepatic stellate cells. FEBS Lett 579: 3119-3125.
-
Stolow MA, Bauzon DD, Li J, Sedgwick T, Liang VC, et al. (1996) Identification and characterization of a novel collagenase in Xenopus laevis: possible roles during frog development. Mol Biol Cell 7: 1471-1483.
-
Patterson ML, Atkinson SJ, Knauper V, Murphy G (2001) Specific collagenolysis by gelatinase A, MMP-2, is determined by the hemopexin domain and not the fibronectin-like domain. FEBS Lett 503: 158-162.
-
Brew K, Dinakarpandian D, Nagase H (2000) Tissue inhibitors of metalloproteinases: evolution, structure and function. Biochim Biophys Acta 1477: 267-283.
-
Murphy G, Houbrechts A, Cockett MI, Williamson RA, O'Shea M, et al. (1991) The N-terminal domain of tissue inhibitor of metalloproteinases retains metalloproteinase inhibitory activity. Biochemistry 30: 8097-8102.
-
Williamson RA, Marston FA, Angal S, Koklitis P, Panico M, et al. (1990) Disulphide bond assignment in human tissue inhibitor of metalloproteinases (TIMP). Biochem J 268: 267-274.
-
Gomis-Ruth FX, Maskos K, Betz M, Bergner A, Huber R, et al. (1997) Mechanism of inhibition of the human matrix metalloproteinase stromelysin-1 by TIMP-1. Nature 389: 77-81.
-
Will H, Atkinson SJ, Butler GS, Smith B, Murphy G (1996) The soluble catalytic domain of membrane type 1 matrix metalloproteinase cleaves the propeptide of progelatinase A and initiates autoproteolytic activation: regulation by TIMP-2 and TIMP-3. J Biol Chem 271: 17119-17123.
-
Meijer MJ, Mieremet-Ooms MA, van der Zon AM, van Duijn W, van Hogezand RA, et al. (2007) Increased mucosal matrix metalloproteinase-1, -2, -3 and -9 activity in patients with inflammatory bowel disease and the relation with Crohn's disease phenotype. Dig Liver Dis 39: 733-739.
-
Arihiro S, Ohtani H, Hiwatashi N, Torii A, Sorsa T, et al. (2001) Vascular smooth muscle cells and pericytes express MMP-1, MMP-9, TIMP-1 and type I protocollagen in inflammatory bowel disease. Histopathology 39: 50-59.
-
Saarialho-Kere UK, Vaalamo M, Puolakkainen P, Airola K, Parks WC, et al. (1996) Enhanced expression of matrilysin, collagenase, and stromelysin-1 in gastrointestinal ulcers. Am J Pathol 148: 519-526.
-
Stumpf M, Cao W, Klinge U, Klosterhalfen B, Junge K, et al. (2005) Reduced expression of collagen type I and increased expression of matrix metalloproteinases 1 in patients with Crohn's disease. J Invest Surg 18: 33-38.
-
Wang YD, Tan XY, Zhang K (2009) Correlation of plasma MMP-1 and TIMP-1 levels and the colonic mucosa expressions in patients with ulcerative colitis. Mediators Inflamm 2009: 275072.
-
Wang YD, Yan PY (2006) Expression of matrix metalloproteinase-1 and tissue inhibitor of metalloproteinase-1 in ulcerative colitis. World J Gastroenterol 12: 6050-6053.
-
Wang H, Keiser JA (1998) Vascular endothelial growth factor upregulates the expression of matrix metalloproteinases in vascular smooth muscle cells: role of flt-1. Circ Res 83: 832-840.
-
Di Sebastiano P, di Mola FF, Artese L, Rossi C, Mascetta G, et al. (2001) Beneficial effects of Batimastat (BB-94), a matrix metalloproteinase inhibitor, in rat experimental colitis. Digestion 63: 234-239.
-
Wiercinska-Drapalo A, Jaroszewicz J, Flisiak R, Prokopowicz D (2003) Plasma matrix metalloproteinase-1 and tissue inhibitor of metalloproteinase-1 as biomarkers of ulcerative colitis activity. World J Gastroenterol 9: 2843-2845.
-
Vaalamo M, Karjialainen-Lindsberg ML, Puolakkainen P, Kere J, Saarialho-Kere U (1998) Distinct expression profiles of stromalysin-2 (MMP10), collagenase-3 (MMP-13), macrophage metalloelastase (MMP-12), and tissue inhibitor of metalloproteinase-3 (TIMP-3) in intestinal ulcerations. Am J Pathol 152: 1005-1014.
-
Becker-Pauly C, Rose-John S (2013) TNFα cleavage beyond TACE/ADAM17: matrix metalloproteinase 13 is a potential therapeutic target in sepsis and colitis. EMBO Mol Med 5: 970-972.
-
Vandenbroucke RE, Dejonckheere E, Van Hauwermeiren F, Lodens S, De Rycke R, et al. (2013) Matrix metalloproteinase 13 modulates intestinal epithelial barrier integrity in inflammatory diseases by activating TNF. EMBO Mol Med 5: 1000-1016.
-
Kar S, Subbaram S, Carrico PM, Melendez JA (2010) Redox-control of matrix metalloproteinase-1: a critical link between free radicals, matrix remodeling and degenerative disease. Respir Physiol Neurobiol 174: 299-306.
-
Wenk J, Brenneisen P, Wlaschek M, Poswig A, Briviba K, et al. (1999) Stable overexpression of manganese superoxide dismutase in mitochondria identifies hydrogen peroxide as a major oxidant in the AP-1-mediated induction of matrix-degrading metalloprotease-1. J Biol Chem 274: 25869-25876.
-
Portal-Nunez S, Esbrit P, Alcaraz MJ, Largo R (2016) Oxidative stress, autophagy, epigenetic changes and regulation by miRNAs as potential therapeutic targets in osteoarthritis. Biochem Pharmacol 108: 1-10.
-
Nakamuta M, Higashi N, Kohjima M, Fukushima M, Ohta S, et al. (2005) Epigallocatechin-3-gallate, a polyphenol component of green tea, suppresses both collagen production and collagenase activity in hepatic stellate cells. Int J Mol Med 16: 677-681.
-
Tyagi SC, Kumar S, Borders S (1996) Reduction-oxidation (redox) state regulation of extracellular matrix metalloproteinases and tissue inhibitors in cardiac normal and transformed fibroblast cells. J Cell Biochem 61: 139-151.
-
Orbe J, Rodriguez JA, Arias R, Belzunce M, Nespereira B, et al. (2003) Antioxidant vitamins increase the collagen content and reduce MMP-1 in a porcine model of atherosclerosis: implications for plaque stabilization. Atherosclerosis 167: 45-53.
-
Svajger U, Jeras M (2012) Anti-inflammatory effects of resveratrol and its potential use in therapy of immune-mediated diseases. Int Rev Immunol 31: 202-222.
-
Moon MH, Jeong JK, Lee YJ, Seol JW, Jackson CJ, et al. (2013) SIRT1, a class III histone deacetylase, regulates TNF-a-induced inflammation in human chondrocytes. Osteoarthritis Cartilage 21: 470-480.
-
Wang ZM, Chen YC, Wang DP (2016) Resveratrol, a natural antioxidant, protects monosodium iodoacetate-induced osteoarthritic pain in rats. Biomed Pharmacother 83: 763-770.
-
Kirkegaard T, Hansen A, Bruun E, Brynskov J (2004) Expression and localisation of matrix metalloproteinases and their natural inhibitors in fistulae of patients with Crohn's disease. Gut 53: 701-709.
-
Dignass AU, Herrlinger K, Scholmerich J (2004) [Ulcerative colitis. Chronic active course]. Z Gastroenterol 42: 1006-1010.
-
Clayburgh DR, Shen L, Turner JR (2004) A porous defense: the leaky epithelial barrier in intestinal disease. Laboratory Investigation 84: 282-291.
-
Garg P, Rojas M, Ravi A, Bockbrader K, Epstein S, et al. (2006) Selective ablation of matrix metalloproteinase-2 exacerbates experimental colitis: contrasting role of gelatinases in the pathogenesis of colitis. J Immunol 177: 4103-4112.
-
Wu X, Yan Q, Zhang Z, Du G, Wan X (2012) Acrp30 inhibits leptin-induced metastasis by downregulating the JAK/STAT3 pathway via AMPK activation in aggressive SPEC-2 endometrial cancer cells. Oncol Rep 27: 1488-1496.
-
Garg P, Vijay-Kumar M, Wang L, Gewirtz AT, Merlin D, et al. (2009) Matrix metalloproteinase-9-mediated tissue injury overrides the protective effect of matrix metalloproteinase-2 during colitis. Am J Physiol Gastrointest Liver Physiol 296: G175-G184.
-
Baugh MD, Evans GS, Hollander AP, Davies DR, Perry MJ, et al. (1998) Expression of matrix metalloproteases in inflammatory bowel disease. Ann N Y Acad Sci 859: 249-253.
-
Bailey CJ, Hembry RM, Alexander A, Irving MH, Grant ME, et al. (1994) Distribution of the matrix metalloproteinases stromalysin, gelatinases A and B, and collagenase in Crohn disease and normal intestine. J Clin Pathol 47: 113-116.
-
Castaneda FE, Walia B, Vijay-Kumar M, Patel NR, Roser S, et al. (2005) Targeted deletion of metalloproteinase 9 attenuates experimental colitis in mice: central role of epithelial-derived MMP. Gastroenterology 129: 1991-2008.
-
Medina C, Radomski MW (2006) Role of matrix metalloproteinases in intestinal inflammation. J Pharmacol Exp Ther 318: 933-938.
-
Fini ME, Parks WC, Rinehart WB, Girard MT, Matsubara M, et al. (1996) Role of matrix metalloproteinases in failure to re-epithelialize after corneal injury. Am J Pathol 149: 1287-1302.
-
Mohan R, Chintala SK, Jung JC, Villar WV, McCabe F, et al. (2002) Matrix metalloproteinase gelatinase B (MMP-9) coordinates and effects epithelial regeneration. J Biol Chem 277: 2065-2072.
-
Behzadian MA, Wang XL, Windsor LJ, Ghaly N, Caldwell RB (2001) TGF-beta increases retinal endothelial cell permeability by increasing MMP-9: possible role of glial cells in endothelial barrier function. Invest Ophthalmol Vis Sci 42: 853-859.
-
Alexander JS, Sasaki M (2002) Is the gastric epithelial barrier regulated by cadherin recruitment? Eur J Gastroenterol Hepatol 14: 1293-1294.
-
Opdenakker G, Van den Steen PE, Dubois B, Nelissen I, Van Coillie E, et al. (2001) Gelatinase B functions as regulator and effector in leukocyte biology. J Leukoc Biol 69: 851-859.
-
Atkinson JJ, Senior RM (2003) Matrix metalloproteinase-9 in lung remodeling. Am J Respir Cell Mol Biol 28: 12-24.
-
Scott KA, Arnott CH, Robinson SC, Moore RJ, Thompson RG, et al. (2004) TNF-a regulates epithelial expression of MMP-9 and integrin avb6 during tumour promotion. A role for TNF-a in keratinocyte migration? Oncogene 23: 6954-6966.
-
Lakatos G, Hritz I, Varga MZ, Juhasz M, Miheller P, et al. (2012) The impact of matrix metalloproteinases and their tissue inhibitors in inflammatory bowel diseases. Dig Dis 30: 289-295.
-
Matusiewicz M, Neubauer K, Mierzchala-Pasierb M, Gamian A, Krzystek-Korpacka M (2014) Matrix metalloproteinase-9: its interplay with angiogenic factors in inflammatory bowel diseases. Dis Markers 2014: 643645.
-
Kofla-Dlubacz A, Matusiewicz M, Krzystek-Korpacka M, Iwanczak B (2012) Correlation of MMP-3 and MMP-9 with Crohn's disease activity in children. Dig Dis Sci 57: 706-712.
-
Kofla-Dlubacz A, Matusiewicz M, Krzesiek E, Noga L, Iwanczak B (2014) Metalloproteinase-3 and -9 as novel markers in the evaluation of ulcerative colitis activity in children. Adv Clin Exp Med 23: 103-110.
-
Farkas K, Sarodi Z, Balint A, Foldesi I, Tiszlavicz L, et al. (2015) The diagnostic value of a new fecal marker, matrix metalloprotease-9, in different types of inflammatory bowel diseases. J Crohns Colitis 9: 231-217.
-
Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, et al. (2000) Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol 2: 737-744.
-
Chantrain CF, Shimada H, Jodele S, Groshen S, Ye W, et al. (2004) Stromal matrix metalloproteinase-9 regulates the vascular architecture in neuroblastoma by promoting pericyte recruitment. Cancer Res 64: 1675-1686.
-
Algieri F, Rodriguez-Nogales A, Vezza T, Garrido-Mesa J, Garrido-Mesa N, et al. (2016) Anti-inflammatory activity of hydroalcoholic extracts of Lavandula dentata L. and Lavandula stoechas L. J Ethnopharmacol 190: 142-158.
-
Te Velde AA, de Kort F, Sterrenburg E, Pronk I, ten Kate FJ, et al. (2007) Comparative analysis of colonic gene expression of three experimental colitis models mimicking inflammatory bowel disease. Inflamm Bowel Dis 13: 325-330.
-
Daum S, Bauer U, Foss HD, Schuppan D, Stein H, et al. (1999) Increased expression of mRNA for matrix metalloproteinases-1 and -3 and tissue inhibitor of metalloproteinases-1 in intestinal biopsy specimens from patients with coeliac disease. Gut 44: 17-25.
-
Heuschkel RB, MacDonald TT, Monteleone G, Bajaj-Elliott M, Smith JA, et al. (2000) Imbalance of stromelysin-1 and TIMP-1 in the mucosal lesions of children with inflammatory bowel disease. Gut 47: 57-62.
-
Pender SL, Tickle SP, Docherty AJ, Howie D, Wathen NC, et al. (1997) A major role for matrix metalloproteinases in T cell injury in the gut. J Immunol 158: 1582-1590.
-
Pender SLF, Fell JME, Chamow SM, Ashkenazi A, MacDonald TT, et al. (1998) A p55 TNF receptor immunoadhesin prevents T cell-mediated intestinal injury by inhibiting matrix metalloproteinase production. J Immunol 160: 4098-4103.
-
Salmela MT, MacDonald TT, Black D, Irvine B, Zhuma T, et al. (2002) Upregulation of matrix metalloproteinases in a model of T cell mediated tissue injury in the gut: analysis by gene array and in situ hybridisation. Gut 51: 540-547.
-
Gupta SC, Patchva S, Aggarwal BB (2013) Therapeutic roles of curcumin: lessons learned from clinical trials. AAPS J 15: 195-218.
-
Shishodia S (2013) Molecular mechanisms of curcumin action: gene expression. Biofactors 39: 37-55.
-
Epstein J, Docena G, MacDonald TT, Sanderson IR (2010) Curcumin suppresses p38 mitogen-activated protein kinase activation, reduces IL-1beta and matrix metalloproteinase-3 and enhances IL-10 in the mucosa of children and adults with inflammatory bowel disease. Br J Nutr 103: 824-832.
-
Gill SE, Parks WC (2008) Metalloproteinases and their inhibitors: regulators of wound healing. Int J Biochem Cell Biol 40: 1334-1347.
-
Wielockx B, Libert C, Wilson C (2004) Matrilysin (matrix metalloproteinase-7): a new promising drug target in cancer inflammation. Cytokine Growth Factor Rev 15: 111-115.
-
Gaire M, Magbanua Z, McDonnell S, McNeil L, Lovett DH, et al. (1994) Structure and expression of the human gene for the matrix metalloproteinase matrilysin. J Biol Chem 269: 2032-2040.
-
McDonnell S, Navre M, Coffey RJ Jr, Matrisian LM (1991) Expression and localization of the matrix metalloproteinase pump-1 (MMP-7) in human gastric and colon carcinomas. Mol Carcinog 4: 527-533.
-
Yoshimoto M, Itoh F, Yamamoto H, Hinoda Y, Imai K, et al. (1993) Expression of MMP-7(PUMP-1) mRNA in human colorectal cancers. Int J Cancer 54: 614-618.
-
Matsuno K, Adachi Y, Yamamoto H, Goto A, Arimura Y, et al. (2003) The expression of matrix metalloproteinase matrilysin indicates the degree of inflammation in ulcerative colitis. J Gastroenteral 38: 348-354.
-
Salmela MT, Pender SLF, Karjalainen-Lindsberg ML, Puolakkainen P, MacDonald TT, et al. (2004) Collagenase-1 (MMP-1), matrilysin-1 (MMP-7), and stromelysin-2 (MMP-10) are expressed by migrating enterocytes during intestinal wound healing. Scand J Gastroenteral 39: 1095-1104.
-
Jakubowska K, Pryczynicz A, Iwanowicz P, Niewinski A, Maciorkowska E, et al. (2016) Expressions of Matrix Metalloproteinases (MMP-2, MMP-7, and MMP-9) and Their Inhibitors (TIMP-1, TIMP-2) in Inflammatory Bowel Diseases. Gastroenterol Res Pract 2016: 2456179.
-
Makitalo L, Sipponen T, Karkkainen P, Kolho KL, Saarialho-Kere U (2009) Changes in matrix metalloproteinase (MMP) and tissue inhibitors of metalloproteinases (TIMP) expression profile in Crohn's disease after immunosuppressive treatment correlate with histological score and calprotectin values. Int J Colorectal Dis 24: 1157-1167.
-
Bister VO, Salmela MT, Karjalainen-Lindsberg ML, Uria J, Lohi J, et al. (2004) Differential expression of three matrix metalloproteinases, MMP-19, MMP-26 and MMP-28, in normal and inflamed intestine and colon cancer. Dig Dis Sci 49: 653-661.
-
Pender SL, Li CK, Di Sabatino A, MacDonald TT, Buckley MG (2006) Role of macrophage metalloelastase in gut inflammation. Ann N Y Acad Sci 1072: 386-388.
-
Brauer R, Tureckova J, Kanchev I, Khoylou M, Skarda J, et al. (2016) MMP-19 deficiency causes aggravation of colitis due to defects in innate immune cell function. Mucosal Immunol 9: 974-985.
-
Rath T, Roderfeld M, Halwe JM, Tschuschner A, Roeb E, et al. (2010) Cellular sources of MMP-7, MMP-13 and MMP-28 in ulcerative colitis. Scand J Gastroenterol 45: 1186-1196.
-
Moss ML, Sklair-Tavron L, Nudelman R (2008) Drug insight: tumor necrosis factor-converting enzyme as a pharmaceutical target for rheumatoid arthritis. Nat Clin Pract Rheumatol 4: 300-309.
-
Alexopoulou L, Kranidioti K, Xanthoulea S, Denis M, Kotanidou A, et al. (2006) Transmembrane TNF protects mutant mice against intracellular bacterial infections, chronic inflammation and autoimmunity. Eur J Immunol 36: 2768-2780.
-
Nowell MA, Williams AS, Carty SA, Scheller J, Hayes AJ, et al. (2009) Therapeutic targeting of IL-6 trans signaling counteracts STAT3 control of experimental inflammatory arthritis. J Immunol 182: 613-622.
-
Lo CW, Chen MW, Hsiao M, Wang S, Chen Chen CA, (2011) IL-6 trans-signaling in formation and progression of malignant ascites in ovarian cancer. Cancer Res 71: 424-434.
-
Xu P, Derynck R (2010) Direct activation of TACE-mediated ectodomain shedding by p38 MAP kinase regulates EGF receptor-dependent cell proliferation. Mol Cell 37: 551-566.
-
Wang Y, Herrera AH, Li Y, Belani KK, Walcheck B (2009) Regulation of mature ADAM17 by redox agents for L-selectin shedding. J Immunol 182: 2449-2457.
-
Zeng SY, Chen X, Chen SR, Li Q, Wang YH, et al. (2013) Upregulation of Nox4 promotes angiotensin II-induced epidermal growth factor receptor activation and subsequent cardiac hypertrophy by increasing ADAM17 expression. Can J Cardiol 29: 1310-1319.
-
Fontani F, Domazetovic V, Marcucci T, Vincenzini MT, Iantomasi T, et al. (2016) Tumor Necrosis Factor-Alpha Up-Regulates ICAM-1 Expression and Release in Intestinal Myofibroblasts by Redox-Dependent and -Independent Mechanisms. J Cell Biochem 117: 370-381.
-
Slomiany BL, Slomiany A (2016) Helicobacter pylori-induced gastric mucosal TGF-a ectodomain shedding and EGFR transactivation involves Rac1/p38 MAPK-dependent TACE activation. Inflammopharmacology 24: 23-31.
-
Urriola-Munoz P, Lagos-Cabre R, Moreno RD (2014) A mechanism of male germ cell apoptosis induced by bisphenol-A and nonylphenol involving ADAM17 and p38 MAPK activation. PLoS One 9: e113793.
-
Ma R, Gu B, Gu Y, Groome LJ, Wang Y (2014) Down-regulation of TIMP3 leads to increase in TACE expression and TNFα production by placental trophoblast cells. Am J Reprod Immunol 71: 427-433.
-
Tsukamoto S, Takeuchi M, Kawaguchi T, Togasaki E, Yamazaki A, et al. (2014) Tetraspanin CD9 modulates ADAM17-mediated shedding of LR11 in leukocytes. Exp Mol Med 46: e89.
-
Bzowska M, Jura N, Lassak A, Black RA, Bereta J, et al. (2004) Tumor necrosis factor-alpha stimulates expression of TNF-alpha converting enzyme in endothelial cells. Eur J Biochem. 271: 2808-2820.
-
Betis F, Brest P, Hofman V, Guignot J, Kansau I, et al. (2003) Afa/Dr diffusely adhering Escherichia coli infection in T84 cell monolayers induces increased neutrophil transepithelial migration, which in turn promotes cytokine-dependent upregulation of decay-accelerating factor (CD55), the recptor for afa/Dr adhesins. Infect Immun 71: 1774-1783.
-
Cesaro A, Abakar-Mahamat A, Brest P, Lassalle S, Selva E, et al. (2009) Differential expression and regulation of ADAM17 and TIMP3 in acute inflamed intestinal epithelia. Am J Physiol Gastrointest Liver Physiol 296: G1332-G1343.
-
Chalaris A, Adam N, Sina C, Rosenstiel P, Lehmann-Koch J, et al. (2010) Critical role of the disintegrin metalloprotease ADAM17 for intestinal inflammation and regeneration in mice. J Exp Med 207: 1617-1624.
-
Shimoda M, Horiuchi K, Sasaki A, Tsukamoto T, Okabayashi K, et al. (2016) Epithelial Cell-Derived a Disintegrin and Metalloproteinase-17 Confers Resistance to Colonic Inflammation Through EGFR Activation. EBioMedicine 5: 114-124.
-
Freour T, Jarry A, Bach-Ngohou K, Dejoie T, Bou-Hanna C, et al. (2009) TACE inhibition amplifies TNF-alpha-mediated colonic epithelial barrier disruption. Int J Mol Med 23: 41-48.
-
Monteleone I, Federici M, Sarra M, Franze E, Casagrande V, et al. (2012) Tissue Inhibitor of Metalloproteinase-3 regulates inflammation in human and mouse intestine. Gastroenterology 143: 1277-1287.
-
Sharma M, Mohapatra J, Wagh A, Patel HM, Pandey D, et al. (2014) Involvement of TACE in colon inflammation: a novel mechanism of regulation via SIRT-1 activation. Cytokine 66: 30-39.
-
DasGupta S, Murumkar PR, Giridhar R, Yadav MR (2009) Studies on novel 2-imidazolidinones and tetrahydropyrimidin-2(1H)-ones as potential TACE inhibitors: design, synthesis, molecular modeling, and preliminary biological evaluation. Bioorg Med Chem 17: 3604-3617.
-
Franze E, Caruso R, Stolfi C, Sarra M, Cupi ML, et al. (2013) High expression of the "A Disintegrin And Metalloprotease" 19 (ADAM19), a sheddase for TNF-α in the mucosa of patients with inflammatory bowel diseases. Inflamm Bowel Dis 19: 501-511.
-
Loui, E, Ribbens C, Godon A, D Franchimont, D De Groote, et al. (2000) Increased production of matrix metalloproteinase-3 and tissue inhibitor of metalloproteinase-1 by inflamed mucosa in inflammatory bowel disease. Clin Exp Immunol 120: 241–246.
-
McKaig BC, McWilliams D, Watson SA, Mahida YR (2003) Expression and regulation of tissue inhibitor of metalloproteinase-1 and matrix metalloproteinases by intestinal myofibroblasts in infiammatory bowel disease. Am J Pathol 162: 1355-1360.
-
Di Sabatino A, Jackson CL, Pickard KM, Buckley M, Rovedatti L, et al. (2009) Transforming growth factor beta signalling and matrix metalloproteinases in the mucosa overlying Crohn's disease strictures. Gut 58: 777-789.
-
Kapsoritakis AN, Kapsoritaki AI, Davidi IP, Lotis VD, Manolakis AC, et al. (2008) Imbalance of tissue inhibitors of metalloproteinases (TIMP) -1 and -4 serum levels, in patients with inflammatory bowel disease. BMC Gastroenterol 8: 55.
-
Varga Z, Herszenyi L, Hritz I (2011) The behavior of serum MMP-2, MMP-7, MMP-9, TIMP-1 AND TIMP-2 concentrations in inflammatory bowel diseases. Z Gastroenterol 49: A93.
-
Smookler DS, Mohammed FF, Kassiri Z, Duncan GS, Mak TW, et al. (2006) Tissue inhibitor of metalloproteinase 3 regulates TNF-dependent systemic inflammation. J Immunol 176: 721-725.
-
Chen M, Peyrin-Biroulet L, George A, Coste F, Bressenot A, et al. (2011) Methyl deficient diet aggravates experimental colitis in rats. J Cell Mol Med 15: 2486-2497.
-
Navasa N, Martín I, Iglesias-Pedraz JM, Beraza N, Atondo E, et al. (2015) Regulation of oxidative stress by methylation-controlled J protein controls macrophage responses to inflammatory insults. J Infect Dis. 211: 135-145.
-
Li WQ, Qureshi HY, Liacini A, Dehnade F, Zafarullah M (2004) Transforming growth factor Beta1 induction of tissue inhibitor of metalloproteinases 3 in articular chondrocytes is mediated by reactive oxygen species. Free Radic Biol Med 37: 196-207.
-
Radomski A, Jurasz P, Sanders EJ, Overall CM, Bigg HF, et al. (2002) Identification, regulation and role of tissue inhibitor of metalloproteinases-4 (TIMP-4) in human platelets. Br J Pharmacol 137: 1330-1338.
-
Andoh A, Yoshida T, Yagi Y, Bamba S, Hata K, et al. (2006) Increased aggregation response of platelets in patients with inflammatory bowel disease. J Gastroenterol 41: 47-54.
