

International Journal of Virology and AIDS
Liver Fibrosis in HIV and HCV Co-Infection: Independent Mechanisms versus Collective Efforts
Jose D Debes1*, Paul R Bohjanen1 and Andre Boonstra2
1Department of Medicine, University of Minnesota, USA
2Department of Gastroenterology and Hepatology, Erasmus MC, University Medical Center, The Netherlands
*Corresponding author: Jose D Debes, MD, Department of Medicine, University of Minnesota, 420 SE Delaware St, Minneapolis, MN, 55455, USA, Tel: +16126256999, E-mail: Debes003@umn.edu
Int J Virol AIDS, IJVA-2-008, (Volume 2, Issue 1), Review Article; ISSN: 2469-567X
Received: April 03, 2015 | Accepted: May 25, 2015 | Published: May 27, 2015
Citation: Debes JD, Bohjanen PR, Boonstra A (2015) Liver Fibrosis in HIV and HCV Co-Infection: Independent Mechanisms versus Collective Efforts. Int J Virol AIDS 2:008. 10.23937/2469-567X/1510008
Copyright: © 2015 Debes JD, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Liver disease is the leading cause of death not related to AIDS in patients infected with human Immunodeficiency Virus (HIV). Patients co-infected with HIV and hepatitis C virus (HCV) experience a more rapid progression liver to fibrosis and develop hepatocellular carcinoma at a younger age than those infected with either virus alone. The mechanisms promoting this accelerated progression to fibrosis are not totally understood. Moreover, it is unclear whether both viruses exert fibrosis progression via independent paths or acting in synergy. The proposed mechanisms for rapid fibrosis progression include, but are not limited to, production of reactive oxygen species following hepatocyte infection by HCV, expression of inflammatory cytokines by Kupffer cells, mitochondrial toxicity, and infection of hepatic stellate cells by HIV, increased bacterial translocation, and modulation of complex immune states in the liver. In the following review we describe the most relevant suggested mechanisms of accelerated fibrosis in HIV/HCV co-infection and we discuss the evidence behind these proposed mechanisms.
Keywords
Liver fibrosis, HIV, HCV, Immunity
Liver Fibrosis
Worldwide, approximately 10-25% of human immunodeficiency virus (HIV) infected people are also chronically infected with the hepatitis C virus (HCV) [1]. Since the introduction of antiretroviral therapy, life expectancy of HIV-infected patients has dramatically improved. As a consequence, HCV-related end-stage liver disease is presently an important cause of morbidity and the second-most important cause of death after HIV-related mortality in co-infected patients [1,2]. Patients co-infected with HCV and HIV has increased mortality rates compared to those with HCV or HIV infection alone. Interestingly, co-infected patients develop hepatocellular carcinoma at a younger age than HCV-monoinfected patients, and suffer an accelerated rate of progression to fibrosis [3]. It is widely understood that HIV monoinfection can contribute to liver fibrosis by multiple general mechanisms such as increase in bacterial translocation from the gut, increased mitochondrial toxicity and direct cytopathic effect, among others. However, it is unclear whether both viruses increase fibrosis progression via independent mechanisms, or act in conjunction, potentiating each other through synergistic pathways.
Hepatic fibrosis is a dynamic process initiated by liver injury that results in increased deposition of extra cellular matrix in the space of Disse, the area in between the hepatocytes and the liver sinusoids, mainly inhabited by hepatic stellate cells (HSC). Eventually, accumulation of extracellular matrix proteins and their decreased removal by matrix metalloproteinases results in a progressive replacement of the liver parenchyma by scar tissue, leading to liver cirrhosis and its complications [4]. The production of reactive oxygen species (ROS) is one of the main triggers for liver injury that results in the cascade that leads to fibrosis. ROS are oxygen-containing free radicals that cause injury through oxidation of lipids and proteins. Dysregulation of mitochondria in the liver is the most recognized source of ROS during hepatic injury [5]. Other stimuli include lipid peroxides and inflammatory cytokines, all of which will eventually lead to activation of HSCs with subsequent extracellular matrix accumulation and fibrosis [6]. In addition, recent studies suggest that autophagy, a metabolic process in which cells digest their own organelles, activate HSC, initiating and stimulating liver fibrosis [7].
The Role of HCV
Chronic infection with HCV is characterized by progression to liver fibrosis, resulting in cirrhosis and/or hepatocellular carcinoma. When HCV infects hepatocytes its core protein can modulate the mitochondrial membrane to induce ROS [8]. In cellular models, the HCV non-structural protein NS5A modulates calcium homeostasis altering the concentration of cytosolic calcium and inducing oxidative stress with production of ROS [9]. This increase in ROS results in fatty deposition in the sinusoids and eventual fibrosis. HCV-infected hepatocytes induce immune activation in the liver, with production of TNF-alpha and IL-1, which leads to activation of HSCs and creates an imbalance between matrix metalloproteinases and tissue-inhibitor metalloproteinases. Moreover, once HCV infection is established, HCV can exert modulation of chemokines grandients in the liver, which affects chemotaxis of immune cells into the liver. Chemokines, such as CCR2 can promote recruitment of inflammatory macrophages into the sinusoids, thus increasing progression to liver injury [10]. Because of the role of chemokines in liver fibrosis, they have become attractive targets for treatment of liver fibrosis. For example, CCR5 targeting is currently being developed as antifibrotic therapy for HIV/HCV co-infected patients [11]. Kupffer cells infected with HCV increase expression and release of Tumor Growth Factor Beta (TGF-beta), one of the best-characterized fibrogenic cytokines [12]. TGF-beta stimulates collagen I and alpha-smooth muscle actin, both of which are important components of the extracellular matrix [13]. HCV-Infected hepatocytes also undergo apoptosis. Growing evidence suggests that following intake of apoptotic bodies, Kupffer cells express TNF and the death receptor component, TRAIL, both of which worsens liver injury [14,15].
The Role of HIV
Although HIV does not replicate in hepatocytes, the HIV co-receptors CXCR4 and CCR5 are expressed in the hepatocyte surface, and the HIV protein gp120 can induce cell signaling in the liver through these co-receptors. HIV does, however, infect HSC. In these cells, gp120 induces activation of tissue-inhibitor metalloproteinases and the monocyte chemo-attractant protein 1 with subsequent HSC chemotaxis and accumulation, leading to liver inflammation and fibrogenesis [16,17]. Outside of the liver, HIV infection leads to a substantial decrease of CD4+ T cells in the gut, causing an increased "microbial permeability" in the gastrointestinal mucosa, with bacterial translocation into the portal and systemic circulation [18]. The increase in lipopolysaccharide secondary to bacterial translocation can stimulate Toll-like receptor 4 in the liver, stimulating TGF-beta production and prompting activation of the fibrogenic cascade [19]. Independent from direct viral effects, antiretroviral therapy for HIV, particularly nucleoside reverse transcriptase inhibitors, can induce mitochondrial toxicity. The lack of normal mitochondrial function impairs beta-oxidation of fatty acids with accumulation of these acids in the cellular cytosol leading to liver fibrosis [20]. In addition, HIV-infected patients on antiretroviral therapy, particularly those on protease inhibitors, frequently experience lipodystrophy (known as HIV-associated lipodystrophy syndrome). Protease inhibitors can decrease peripheral lipolysis through inhibition of GLUT-4 activity increasing adipocyte size [21]. These hypertrophic adipocytes in peripheral tissue and abdomen lose functional activity and become resistant to insulin. Consequently, insulin-resistant adipocytes secret less adiponectin, which in turn increases body fat worsening deposition of liver fat and fibrosis.
HIV and HCV Co-Infection
Bacterial translocation
As mentioned above, HIV-infected hosts experience an enhanced bacterial translocation, with elevated levels of circulating lipopolysaccharide leading to an increase state of immune activation. In patients co-infected with HCV and advanced liver disease, there is less ability to clear bacteria from the liver, exacerbating an already increased immune-active state due to HCV infection. This enhanced inflammation creates an environment for more rapid liver fibrosis progression. Interestingly, patients with high HIV viral load, can have depletion of Kupffer cells [22]. On one hand this could worsen liver fibrosis, since Kupffer cells are the most active phagocytes in the liver, therefore "unopposed" lipopolysaccharide from poor bacterial clearance could promote greater fibrosis by HSC. On the other hand, a depletion of Kupffer cells in this setting could lead to decreased phagocytosis of apoptotic hepatocytes and less production of TNF and other inflammatory cytokines, thereby decreasing the severity of fibrosis.
Insulin resistance
HIV patients can present with HIV-associated lipodystrophy syndrome. Moreover, antiretroviral therapy can cause or worsen existing lipodystrophy. This may lead to insulin resistance, which itself can promote fatty liver. High levels of insulin and glucose stimulate HSC proliferation and increase expression of connective tissue growth factor which promotes liver fibrosis progression [6]. Insulin resistance is a well-defined pathological feature of HCV infection. Co-infection with HIV or treatment of HIV with protease inhibitors in co-infected patients could further increase insulin resistance. It is possible that upon control of HIV infection with protease inhibitors, co-infected patients might experience a heightened state of insulin resistance, thereby worsening liver injury.
Modulation of CD4+ T lymphocytes
It is well known that the HIV-induced reduction of CD4 counts in the liver has an overall negative impact on the progression of HCV disease. Interestingly, a recent study suggests that CD4+ T cell depletion decreases the anti-fibrotic activity of NK cells in the liver [23]. Reduced CD4 counts result in decreased levels of IFN-alpha, which has antifibrotic activity in HCV-infected patients. A decrease of IFN-alpha levels can certainly promote more rapid liver fibrosis [6]. Indeed, high CD4 counts in HIV/HCV co-infected patients have been associated with a reduced risk of liver fibrosis [24].
Conclusion
The mechanisms leading to rapid liver fibrosis in HIV/HCV co-infected patients are highly complex with immunological, metabolic and viral factors all playing important roles (Figure 1). Many questions remain unanswered about whether HIV and HCV potentiate each other's effect on liver fibrosis through synergistic mechanisms, particularly those related to immunity. In addition to HIV and HCV, other important cofactors can mediate accelerated fibrosis, such as obesity, fatty liver, differences in the microbiome, altered farnesoid x receptor-mediated pathways or polymorphisms in the PNPLA3 gene [3,25,26]. Moreover, common pathways inducing fibrosis in co-infected patients can be worsened upon treatment of HIV in these patients. Independent mechanisms of liver injury coexist during HIV/HCV co-infection that can substantially accelerate liver fibrosis. This diversity of effects that promotes an "unfriendly" liver environment poses a dramatic challenge to understand and halt the development of fibrosis and consequent cirrhosis of the liver. Further research, particularly regarding assessment of inflammation in intra-hepatic samples from co-infected patients before and after initiating treatments, will be necessary to shed some light on this complex interaction.
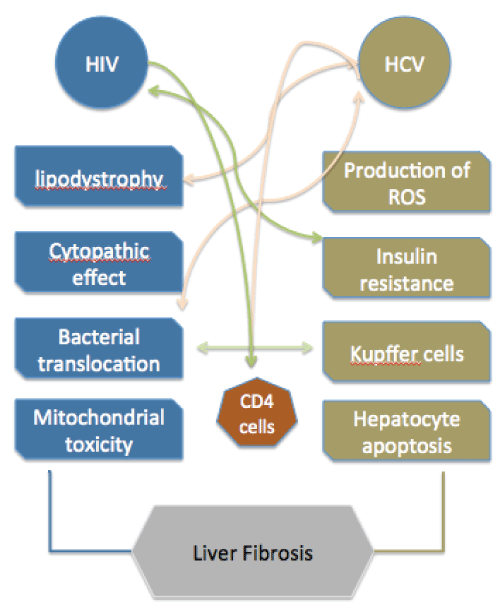
.
Figure 1: Mechanisms of liver fibrosis during HIV/HCV co-infection and points of interaction.
View Figure 1
Acknowledgment
AB is partially supported by Virgo consortium, funded by the Dutch government project number FES0908. PRB is partially supported by grant K24AI096925 from the NIH
References
-
Joshi D, O'Grady J, Dieterich D, Gazzard B, Agarwal K (2011) Increasing burden of liver disease in patients with HIV infection. Lancet 377: 1198-1209.
-
Chen JY, Feeney ER, Chung RT (2014) HCV and HIV co-infection: mechanisms and management. Nat Rev Gastroenterol Hepatol 11: 362-371.
-
Price JC, Seaberg EC, Latanich R, Budoff MJ, Kingsley LA, et al. (2014) Risk factors for fatty liver in the Multicenter AIDS Cohort Study. Am J Gastroenterol 109: 695-704.
-
Singh S, Allen AM, Wang Z, Prokop LJ, Murad MH, (2015) Fibrosis Progression in Nonalcoholic Fatty Liver vs Nonalcoholic Steatohepatitis: A Systematic Review and Meta-analysis of Paired-Biopsy Studies. Clin Gastroenterol Hepatol 13:643-54.e9.
-
Lin W, Weinberg EM, Chung RT (2013) Pathogenesis of accelerated fibrosis in HIV/HCV co-infection. J Infect Dis 207: S13-18.
-
Mastroianni CM, Lichtner M, Mascia C, Zuccal� P, Vullo V (2014) Molecular mechanisms of liver fibrosis in HIV/HCV coinfection. Int J Mol Sci 15: 9184-9208.
-
Song Y, Zhao Y, Wang F, Tao L, Xiao J, et al. (2014) Autophagy in hepatic fibrosis. Biomed Res Int 2014: 436242.
-
Choi J (2012) Oxidative stress, endogenous antioxidants, alcohol, and hepatitis C: pathogenic interactions and therapeutic considerations. Free Radic Biol Med 52:1135-1150.
-
Dionisio N, Garcia-Mediavilla MV, Sanchez-Campos S, Majano PL, Benedicto I, et al. (2009) Hepatitis C virus NS5A and core proteins induce oxidative stress-mediated calcium signalling alterations in hepatocytes. J Hepatol 50: 872-882.
-
Brass A, Brennd�rfer ED (2014) The role of chemokines in hepatitis C virus-mediated liver disease. Int J Mol Sci 15: 4747-4779.
-
Gonzalez EO, Boix V, Deltoro MG, Aldeguer JL, Portilla J, et al. (2014) The effects of Maraviroc on liver fibrosis in HIV/HCV co-infected patients. J Int AIDS Soc 17: 19643.
-
Taniguchi H, Kato N, Otsuka M, Goto T, Yoshida H, et al. (2004) Hepatitis C virus core protein upregulates transforming growth factor-beta 1 transcription. J Med Virol 72: 52-59.
-
Lin W, Tsai WL, Shao RX, Wu G, Peng LF, et al. (2010) Hepatitis C virus regulates transforming growth factor beta1 production through the generation of reactive oxygen species in a nuclear factor kappaB-dependent manner. Gastroenterology 138: 2509-2518.
-
Yoon JH, Gores GJ (2002) Death receptor-mediated apoptosis and the liver. J Hepatol 37: 400-410.
-
Hosomura N, Kono H, Tsuchiya M, Ishii K, Ogiku M, et al. (2011) HCV-related proteins activate Kupffer cells isolated from human liver tissues. Dig Dis Sci 56: 1057-1064.
-
Tuyama AC, Hong F, Saiman Y, Wang C, Ozkok D, et al. (2010) Human immunodeficiency virus (HIV)-1 infects human hepatic stellate cells and promotes collagen I and monocyte chemoattractant protein-1 expression: implications for the pathogenesis of HIV/hepatitis C virus-induced liver fibrosis. Hepatology 52: 612-622.
-
Bruno R, Galastri S, Sacchi P, Cima S, Caligiuri A, et al. (2010) gp120 modulates the biology of human hepatic stellate cells: a link between HIV infection and liver fibrogenesis. Gut 59: 513-520.
-
Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, et al. (2006) Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med 12: 1365-1371.
-
Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, et al. (2007) TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med 13: 1324-1332.
-
P�rez-Matute P, P�rez-Mart�nez L, Blanco JR, Oteo JA (2013) Role of mitochondria in HIV infection and associated metabolic disorders: focus on nonalcoholic fatty liver disease and lipodystrophy syndrome. Oxid Med Cell Longev 493413.
-
Carper MJ, Cade WT, Cam M, Zhang S, Shalev A, et al. (2008) HIV-protease inhibitors induce expression of suppressor of cytokine signaling-1 in insulin-sensitive tissues and promote insulin resistance and type 2 diabetes mellitus. Am J Physiol Endocrinol Metab 294: E558-567.
-
Balagopal A, Philp FH, Astemborski J, Block TM, Mehta A, et al. (2008) Human immunodeficiency virus-related microbial translocation and progression of hepatitis C. Gastroenterology 135: 226-233.
-
Gl�ssner A, Eisenhardt M, Kokordelis P, Kr�mer B, Wolter F, et al. (2013) Impaired CD4⁺ T cell stimulation of NK cell anti-fibrotic activity may contribute to accelerated liver fibrosis progression in HIV/HCV patients. J Hepatol 59: 427-433.
-
Puoti M, Bonacini M, Spinetti A, Putzolu V, Govindarajan S, et al. (2001) Liver fibrosis progression is related to CD4 cell depletion in patients coinfected with hepatitis C virus and human immunodeficiency virus. J Infect Dis 183: 134-137.
-
Ali AH, Carey EJ, Lindor KD (2015) Recent advances in the development of farnesoid X receptor agonists. Ann Transl Med 3: 5.
-
Kelly P, Saloojee H, Chen JY, Chung RT (2014) Noncommunicable diseases in HIV infection in low- and middle-income countries: gastrointestinal, hepatic, and nutritional aspects. J Acquir Immune Defic Syndr 67: S79-86.
