

International Journal of Respiratory and Pulmonary Medicine
The Role of Histone Deacetylase Inhibitors in Myocardial Infarction
Ping Zeng and Jian Yang*
Department of Cardiology, China Three Gorges University, China
*Corresponding author: Jian Yang, Department of Cardiology, First College of Clinical Medical Sciences, Institute of Cardiovascular Diseases, China Three Gorges University, Yichang 443000, Hubei Province, Yiling Road 183, China, Tel: +86-15586373063, Fax: +86(0717)6482302, E-mail: yangjian@ctgu.edu.cn
Int J Respir Pulm Med, IJRPM-3-054, (Volume 3, Issue 3), Review Article; ISSN: 2378-3516
Received: August 02, 2016 | Accepted: August 22, 2016 | Published: August 25, 2016
Citation: Zeng P, Yang J (2016) The Role of Histone Deacetylase Inhibitors in Myocardial Infarction. Int J Respir Pulm Med 3:054. 10.23937/2378-3516/1410054
Copyright: © 2016 Zeng P, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Myocardial infarction is usually recognized as the final stage of coronary artery stenosis or occlusion in response to coronary atherosclerosis and thrombosis involving dysfunction and activation of resident vascular cells as well as the release of t-PA. As a member of family proteins for deacetylation of core histones in eukaryotic cells, Histone deacetylases are implicated in various biological processes. Accumulating evidence suggest that Histone deacetylases inhibitors regulate the release of t-PA and pro-inflammatory cytokine which can decrease myocardial infract size and preserve cardiac function ultimately. Here, we review the effect of Histone deacetylases inhibitors on the progress of myocardial infarction.
Keywords
Myocardial infarction, Histone deacetylases inhibitors, t-PA, Pro-inflammatory cytokine
Introduction
It is well known that Histone deacetylase (HDAC) and Histone acetyltransferases (HATs) are two opposing family proteins for acetylation of core histones in eukaryotic cells, Histone deacetylases inhibitors (HDACIs) can inhibit the process of histone deacetylation effectively. Based on homologous degree of yeast cells' transcription factors, HDAC can be classified into four groups: (1) Class I HDACs consist of HDAC 1, 2, 3, and 8; (2) Class II HDACs include HDAC 4, 5, 7, and 9; (3) Class III HDACs : SIRT1-7; (4) Class IV: HDAC 11. Zn2+ is a necessary cofactor for class I, II, IV and coenzyme is needed for class III [1]. HDACIs can hinder histone acetylation process by regulating the tightness of intertwined DNA and histones, make DNA tightly bind to histones [2]. Recently, studies have detected that HDACIs were implicated in stimulating t-PA production [3] and inhibiting pro-inflammatory cytokine generation [4]. What's more, it has been proved lately that HDACIs can inhibit lung cancer and breast cancer [5]. Here, we briefly review the effect of HDACIs on the process of MI (myocardial infarction).
Biology of HDACIs
HDACIs are generally defined as an organic compound that inhibits histone acetylation. Recent researches have showed that broad spectrum HDACIs are well-known anti-inflammatory agents which have the ability to reduce vascular inflammation [6]. On the basis of their chemical constitution, HDACIs can be divided into several categories [7]: Short chain fatty acids (butyrate, butyl benzoic acid ester and valproic acid), Hydroxamic acid (TSA and SAHA), Amino phenyl amide (FK-228) and peptide (MS-275, MGCD0103). The four kinds of HDACIs can regulate histone acetylation degree by inhibiting HDACs bind to histone. As HDACs have essential roles in both the development [8] and activation [9] of many immune cell types, including macrophage, HDACIs can inhibit the activation of macrophages effectively. HDACIs were formerly suggested to be an active target for cancer therapy, many HDACs mutations are associated with different human cancers. However, it is now obvious that many HDACIs are implicated in various biological processes, including myocardial infarction. Coronary atherosclerosis is the most common pathological process that leads to myocardial infarction. It is marked by the recruitment of macrophages and other leukocytes such as memory T cells, as well as non-leukocytes, including vascular smooth muscle cells (VSMCs) and it is now considered a chronic inflammatory disease [10]. For example, HDACI can suppress the release of IL-10 and enhance the expression of anti-inflammatory cytokines [11]. T-PA (tissue plasminogen activator) release has been found to be detective in certain conditions associated with coronary atherosclerosis [12], and it is a fact that t-PA expression can be powerfully up-graduated by HDACIs [13]. MKK3 and Akt-1 pathways both play an essential role in HDACIs–induced cardio protection for the first time and it has been proved that HDACI can improve the ventricular function and alleviate the reduction of myocardial infarct [14]. In addition, previous study demonstrated that PPARr signaling pathway is regulated by HDACIs in heart [15]. Moreover, HDACIs induce sdipogenesis and adipocyte differentiation through PPARr signaling pathway [16]. These studies suggested that HDACIs can regulate cardiac metabolism.
HDACIs and Myocardial Infraction
The progression of myocardial infarction is characterized by several steps: coronary artery occlusion, blood flow interrupted. The pathogenesis of myocardial infarction is very complex, and it is often caused by intravascular thrombus formation. The clinical outcomes can be alleviated if the thrombus is rapidly removed by the endogenous fibrinolytic system. Conversely, it will prolong the ischemic time and cause irreversible tissue damage if the thrombus persist, the extent of myocardial damage correlates directly with the extent of LV (left ventricular) remodeling [17]. There are several signaling pathways contributing to attenuating myocardial infarction by HDACIs (As it is showed in the Figure 1). This multifactorial process is attributed to several important aspects, including decreased infarct size, improved ventricular function, prevented cardiac remodeling, improved regenerated myocardium cells and angiogenesis. A growing quantity of studies have identified that HDACIs can ameliorate myocardial infarction through different pathways, suggesting the possibility of HDACIs in influencing the progression of myocardial infarction. It has been demonstrated that inhibition of Class II HDACs can silence fatal gene activation, block cardiac hypertrophy and prevent cardiac remodeling [18]. Marten A Hoeksema, et al. found that macrophage HDAC3 deletion is beneficial in atherosclerotic mice [19]. What's more, HDACIs constitute a major cascade in controlling cardio genesis and promoting the survival of ESCs (embryonic stem cells).
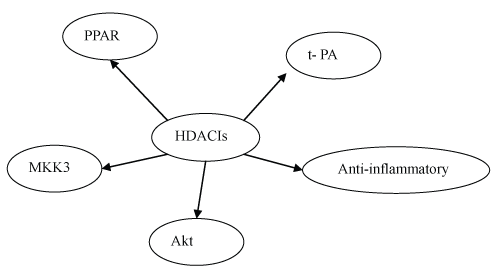
.
Figure 1 HDACIs can attenuate myocardial infarction through different signaling pathways. HDACIs can activate the release of t-PA, exert anti-inflammatory property, and activate Akt, MKK3, and PPAR signaling pathways.
View Figure 1
Roles of HDACIs in activating the release of t-PA
Myocardial infarction is caused by intravascular clot formation, when a clotting process is initiated, the surrounding endothelium is activated and release large amounts of t-PA which can lead the clot to dissolve. t-PA enhancer- 7351C/T which is a low-secretor phenotype was found to be associated with a more than 3-fold adjusted increased risk for myocardial infarction [20]. It has been reported that t-PA release can be powerfully up-regulated by the classical HDACIs, such as butyrate and Trichostatin A (TSA), as well as to the newer HDACI MS-275 [21,22]. It suggests that t-PA gene could be sensitive to the level of histone acetylation status. t-PA is quickly released in the vicinity of the clot [23], what's more, anti-platelet drugs, novel devices (e.g. drug eluting stents), an abiding focus on time to reperfusion have also been proven critical for decreasing infract size and improving cardiac function, and HDACI has promise for fibrinolytic therapy [24].
Roles of HDACIs in exerting anti-inflammatory effect
Recent research shows that broad spectrum HDAC inhibitors are well-known anti-inflammatory agents and can reduce vascular inflammation [25]. Kazuhiro Ito, et al. have demonstrated that theophylline-a kind of HDACIs can decrease inflammatory gene expression [26], while the molecular mechanism for the anti-inflammatory action of theophylline is currently unclear. It has been proved that low concentrations of theophylline are able to inhibit the activation of NF-kB and reduce the expression of inflammatory genes. HDACIs impair the recruitment of nuclear factor NF-kB and IFN regulatory factor-1 which is a counteractive effect of HDAC-dependent IL-12p40 gene in vascular endothelial cell [27]. On the other hand, HDACIs can trigger hyper acetylation of mitogen-activated protein kinase phosphatase-1 which can inhibit LPS-induced mitogen-activated protein kinase p38 (P38MAPK) [28]. Additionally, HDACIs can inhibit the expression of IL-10 which is a counteractive effect of HDAC11 [11].
Roles of HDACIs in Akt and MKK3 signaling pathways
It has been reported that HDAC3 was associated with plaque vulnerability and strongly correlated with macrophage marker CD68, both PPARr and LXR pathways can be upregulated in HDAC3del macrophages and result in less vulnerable lesions [19]. Shiojima I, et al. have demonstrated that Akt serves as a powerful survival signal to protect the heart against myocardial injury [29] and MKK3 has also been proved protective for myocytes [30]. Ting C. Zhao, et al. demonstrated that MKK3 and Akt-1 pathways both play an essential role in HDAC inhibition–induced cardioprotection for the first time and proved that HDACI can improve the ventricular function and alleviate the reduction of myocardial infarct [14]. It is known that AKT will serve as survival signal to protect the heart from myocardial infarction [31]. In addition, the activation of AKT signaling derived from mesenchymal stem cells can catalyse the prevention of cardiac remodeling, and improve the number of regenerated myocardium cells and angiogenesis and restoration of myocardial function [32].
Roles of HDACIs in PPAR signaling pathway
HDACIs can regulate adipogenesis and adipocyte differentiation through PPAR signaling pathway, which suggest the potential of HDACIs on cardiac metabolism [33]. PPAR was widely considered as an anti-inflammatory agent, and it's an important determinant of macrophage polarization, migration, differentiation. HDACIs can inhibit the recruitment of HDACs to the PPAR promoter, thus resulting in enhanced PPAR expression and activity, and suppress inflammation. PPAR-δ has the ability of anti-apoptotic and anti-inflammatory effect, it has showed that PPAR-δ activation inhibits endothelial cell apoptosis and promotes proliferation and angiogenesis after myocardial infarction [34].
Role of HDACIs in the Treatment of Myocardial Infarction
It has been proved that HDACs have a correlation with myocardial infraction in recent years. HDACIs can silence myocytes' fatal gene activation, block cardiac hypertrophy. What's more, HDACIs can protect the heart against myocardial ischemia injury and prevent remodeling. Anne Grange, et al. proved that HDACIs can reduce the size of myocardial infarction between 48.3 and 55.6% depending on different treatment protocols [35]. Ling X. Zhang demonstrated that HDACIs can promote cardio genesis, which is associated with the reduction of HDAC4 [36]. After long-term experiments conducted by different researchers, we can conclude that there are many types of HDACIs which are conducive to ameliorate myocardial infarction. Thus, they may have a therapeutic approach for treating this kind of disease.
Valproic acid (VPA)
VPA is a kind of HDACI which has been proved to increase t-PA expression either in vivo or in vitro, and can also lower plasma levels of plasminogen activator inhibitor-1 (PAI-1). Kristina Svennerholm, et al. did an explorative clinical study among male adult patients with the result showing that HDACI (especially VPA) can stimulate t-PA release, and they also found that VPA pre-treatment could increase t-PA release by a standardized acute ischemic provocation [37]. HDACIs intervention points a promising therapeutic way to improve endogenous fibrinolytic capacity, especially for the patients with high risk of thromboembolic disease. Lower dose of VPA can increase t-PA release, while high dose of VPA can cause manifold increase in it [3]. Olesen, et al. did a research with a result showing that 40% reduced risk for myocardial infarction in the treatment with VPA [38]. Both experiments proved that VPA which function as a HDACI has a positive effect in stimulating t-PA release for thromboembolic disease which can be beneficial for myocardial infarction.
Cyclic phosphatidic acid (CPA)
Cyclic phosphatidic acid (CPA) can inhibit the expression of HDAC 2, and it was isolated for the first time from myxoamoebae of a true slime mold, Physarum polycephalum, in 1992 [39], it is a naturally occurring phospholipid and can be generated by phospholipase D2 (PLD2). CPA consists of a cyclopropane-containing fatty acyl chain and a cyclic phosphate joining the sn-2 and sn-3 positions of glycerol. It has been proved that CPA can regulate peroxisome proliferator-activated receptor gamma (PPAPr) function by stabilizing the silencing mediator of retinoid and thyroid hormone receptors (SMRT)-PPARγ complex [40], and prevent neointima formation, adipocytic differentiation, lipid accumulation [41]. CPA can inhibit the development of atherosclerosis, Tamotsu Tsukahara, et al. did an experiment with a conclusion that CPA can inhibit pro-inflammatory cytokine expression after alkyl-glycerophosphate (AGP) exposed to human coronary artery endothelial cells (HCAECs) [42].
Trichostatin A (TSA)
TSA is a kind of pharmacologic HDACI which can effectively reduce blood pressure and vascular inflammation [43]. HDAC with trichostatin A (TSA) protects the heart against ischemic injury and TSA can stimulate transcription factor kB, gp91, p38 mitogen-activated protein kinase. It has been widely used as a promising anticancer agent. Treat the cultured embryonic stem cells with TSA can stimulates my genesis and angiogenesis which is associated with the restoration/preservation of myocardial function after myocardial infarction, indicating that HDACI can stimulate angiogenesis and thus decrease the size of myocardial infarction after MI [44]. Ting C. Zhao, et al. did an experiment showing that there is a significant greater reduction of myocardial infraction size in TSA treated mice as compared to the control group [24]. What's more, following the treatment of TSA, the trend of LVDP and LVEDP were improved. Ling Zhang, et al. demonstrated that TSA treatment has an amazing effect to increase the content of coronary effluent (CF), and finally concluded that TSA stimulates the self-renewal of c-kit CSCs and enhances endogenous myocardial proliferation and cytokinesis in vivo in MI hearts compared with the control group [34]. They also proved that TSA can improve myocardial functional recovery in the infarcted heart, stimulated endogenous myocardial regeneration, newly formed vascular structure and prevented myocardial remodeling. On the other hand, it has been indicated that TSA pretreatment could ameliorate myocardial damage by the inhibition of CHOP expression and CHOP-induced apoptosis. TSA can inhibit the apoptosis of cardiomyocytes and thus enhance cell viability [45].
Suberoylanilide hydroxamic acid (SAHA)
SAHA is a FDA-approved medicine for treatment of cutaneous T-cell lymphoma, which is a kind of HDACI with the function of anticancer [46]. Min Xie, et al. did an experiment to test the function of both TSA and SAHA in myocardial infraction, adding weight to the significant TSA-dependent protection, what's more, they found that SAHA (50 mg/kg) reduce infarct size by around 45% (p < 0.05), Lower dose of SAHA (30 mg/kg) manifest a trend toward reduced infarct size (§20%) [45]. All of these data established that SAHA is functionally similar to TSA in its ability to blunt I/R damage in mice and rabbit, which is of great therapeutic effects in treating with myocardial infarction [47]. They further discovered that the accumulations of auto phagosomes are observed in the infarct border zone of SAHA-treated, while, SAHA has showed to increase autophagy in cancer cells [48]. Identifying that SAHA's cardio protective effects are dependent on autophagic flux. There are many other mechanisms underlying that SAHA can be cardio protection, for example, SAHA has anti-inflammatory properties and promote the proliferation and homing of stem cells [49]. While, autophagic flux is the primary mechanism for cardio protection.
Script aid and mocetinostat
Script aid is a synthetic HDACI. Script aid resulted in a nearly identical effect when compared to Null script which is a negative control compound for script aid with a 46.8% reduction in infarct size (21.2 ± 3.3%; P = 0.035). These results strongly suggest that in murine models, HDACIs can reverse the induction of ischemia-induced HDAC activity in vivo and reduce myocardial infarct size by more than 50% [35]. Interestingly, pretreatment of hypoxic myocytes with scripted can repress this production to base line without significant change in RNA levers [50]. Mocetinostat is a kind of HDACIs which can inhibit HDAC1, 2, 3, Reseach has showed that the anti-fibrotic mocetinostat can modulate IL-6/STST3 axis and down-regulation of ECM production in myocardium fibroblasts in ischemic heart failure models [51,52]. But more researches should be conducted to demonstrate whether it is effective or not to applying for clinical.
Conclusions and Perspectives
HDACIs have been identified as key regulators in process of several diseases, including myocardial infarction. Numerous researches suggested that HDACIs play critical role in several aspects of myocardial infarction, such as reducing infarct size, improve t-PA level, stimulate angiogenesis, and protect myocardial function (Table 1). Several HDACIs have intensive potentials as biomarkers and therapeutic targets for diverse pathological changes involved in myocardial infarction process. While only a few of them were described to directly contribute to progress of myocardial infarction. However, the functional roles of HDACIs in myocardial infraction requiring further investigation in order to illuminate the pharmacological mechanisms and guide clinical medication in a better way.
![]()
Table 1: HDACIs and their involvements in myocardial infarction.
View Table 1
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 81470387 to J. Yang), and the Natural Science Foundation of Yichang city, China (Grant No.A12301-01) as well as Hubei Province's Outstanding Medical Academic Leader program, China.
References
-
Chen L, Feng Y, Zhou Y, Zhu W, Shen X, et al. (2010) Dural role of Zn 2+ in maintaining structural integrity and suppressing deacetylase activity of SIRT1. J Inorg Biochem 104: 180-185.
-
Ferguson BS, McKinsey TA (2015) Non-sirtuin histone deacetylases in the control of cardiac aging. J Mol Cell Cardiol 83: 14-20.
-
Larsson P, Bergh N, Lu E, Ulfhammer E, Magnusson M, et al. (2013) Histone deacetylase inhibitors stimulate tissue-type plasminogen activator production in vascular endothelial cells. J Thromb Thrombolysis 35: 185-192.
-
Schmitz ML, de la Vega L (2015) New Insights into the Role of Histone Deacetylases as Coactivators of Inflammatory Gene Expression. Antioxid Redox Signal 23: 85-98.
-
Zou Z, Luo X, Nie P, Wu B, Zhang T, et al. (2016) Inhibition of SRC-3 enhances sensitivity of human cancer cells to histone deacetylase inhibitors. Biochem Biophys Res Commun 478: 227-233.
-
Shakespear MR, Halili MA, Irvine KM, Fairlie DP, Sweet MJ (2011) Histone deacetylases as regulators of inflammation and immunity. Trends Immunol 32: 335-343.
-
Bush EW, McKinsey TA (2010) Protein acetylation in the cardiorenal axis: the promise of histone deacetylase inhibitors. Circ Res 106: 272-284.
-
Fairlie DP, Sweet MJ (2012) HDACs and their inhibitors in immunology: teaching anticancer drugs new tricks. Immunol Cell Biol 90: 3-5.
-
Vishwakarma S, Iyer LR, Muley M, Singh PK, Shastry A, et al. (2013) Tubastatin, a selective histone deacetylase 6 inhibitor shows anti-inflammatory and anti-rheumatic effects. Int Immunopharmacol 16: 72-78.
-
Bäck M, Hansson GK (2006) Leukotriene receptors in atherosclerosis. Ann Med 38: 493-502.
-
Villagra A, Cheng F, Wang HW, Suarez I, Glozak M, et al. (2009) The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance. Nat Immunol 10: 92-100.
-
Osterlund B, Jern S, Jern C, Seeman-Lodding H, Ostman M, et al. (2008) Impaired myocardial t-PA release in patients with coronary artery disease. Acta Anaesthesiol Scand 52: 1375-1384.
-
Larsson P, Ulfhammer E, Magnusson M, Bergh N, Lunke S, et al. (2012) Role of Histone Acetylation in the Stimulatory Effect of Valproic Acid on Vascular Endothelial Tissue-Type Plasminogen Activator Expression. Plos One 7: e31573-e31573.
-
Zhao TC, Du J, Zhuang S, Liu P, Zhang LX (2013) HDAC inhibition elicits myocardial protective effect through modulation of MKK3/Akt-1. PLoS One 8: e65474.
-
Lkhagva B, Lin YK, Kao YH,Chazo TF, Chung CC, et al. (2015) Novel Histone Deacetylase Inhibitor Modulates Cardiac Peroxisome Proliferator-Activated Receptors and Inflammatory Cytokines in Heart Failure. Pharmacology 96: 184-191.
-
Nebbioso A, Dell'Aversana C, Bugge A, Sarno R, Valente S, et al. (2010) HDACs class II-selective inhibition alters nuclear receptor-dependent differentiation. J Mol Endocrinol 45: 219-228.
-
Fraccarollo D, Galuppo P, Bauersachs J (2012) Novel therapeutic approaches to post-infarction remodelling. Cardiovasc Res 94: 293-303.
-
Kee HJ, Sohn IS, Nam KI, Park JE, Qian YR, et al. (2006) Inhibition of histone deacetylation blocks cardiac hypertrophy induced by angiotensin II infusion and aortic banding. Circulation 113: 51-59.
-
Hoeksema MA, Gijbels MJ, Bossche JVD, Velden SVD, Sijm A, et al. (2014) Targeting macrophage Histone deacetylase 3 stabilizes atherosclerotic lesions. EMBO Mol Med 6: 1124-1132.
-
Kathiresan S, Yang Q, Larson MG, Camargo AL, Tofler GH, et al. (2006) Common genetic variation in five thrombosis genes and relations to plasma hemostatic protein level and cardiovascular disease risk. Arterioscler Thromb Vasc Biol 26: 1405-1412.
-
Arts J, Lansink M, Grimbergen J, Toet KH, Kooistra T (1995) Stimulation of tissue-type plasminogen activator gene expression by sodium butyrate and trichostatin A in human endothelial cells involves histone acetylation. Biochem J 310: 171-176.
-
Sylvie DG, Kruithof EK (2011) Epigenetic control of tissue-type plasminogen activator synthesis in human endothelial cells. Cardiovasc Res 90: 457-463.
-
Osterlund B, Andersson B, Häggmark S, Jern C, Johansson G, et al. (2002) Myocardial ischemia induces coronary t-PA release in the pig. Acta Anaesthesiol Scand 46: 271-278.
-
Dorn GW (2009) Novel pharmacotherapies to abrogate postinfarction ventricular remodeling. Nat Rev Cardiol 6: 283-291.
-
Huang L (2006) Targeting histone deacetylases for the treatment of cancer and inflammatory diseases. J Cell Physiol 209: 611-616
-
Ito K, Lim S, Caramori G, Cosio B, Chung KF, et al. (2002) A molecular mechanism of action of theophylline: Induction of histone deacetylase activity to decrease inflammatory gene expression. Proc Natl Acad Sci U S A 99: 8921-8926.
-
Bode KA, Kate S, Hume DA, Ravasi T, Heeg K, et al. (2007) Histone deacetylase inhibitors decrease Toll-like receptor-mediated activation of proinflammatory gene expression by impairing transcription factor recruitment. Immunology 122: 596-606.
-
Jeong Y, Du R, Zhu X, Yin S, Wang J, et al. (2014) Histone deacetylase isoforms regulate innate immune responses by deacetylating mitogen-activated protein kinase phosphatase-1. J Leukoc Biol 95: 651-659.
-
Shiojima I, Yefremashvili M, Luo Z, Kureishi Y, Takahashi A, et al. (2002) Akt signaling mediates postnatal heart growth in response to insulin and nutritional status. J Biol Chem 277: 37670-37677.
-
Schulz R, Belosjorow S, Gres P, Jansen J, Michel MC, et al. (2002) p38 MAP kinase is a mediator of ischemic preconditioning in pigs. Cardiovasc Res 55: 690-700.
-
Matsui T, Tao J, Monte F D, et al. (2001) Akt activation preserves cardiac function and prevents injury after transient cardiac ischemia in vivo. Circulation 104: 165-165.
-
Gnecchi M, He H, Liang OD, Melo LG, Morello F, et al. (2005) Paracrine action accounts for marked protection of ischemic heart by Akt-modified mesenchymal stem cells. Nat Med 11: 367-368.
-
Haberland M, Carrer M, Mokalled MH, Montgomery RL, Olson EN (2010) Redundant control of adipogenesis by histone deacetylases 1 and 2. J Biol Chem 285: 14663-14670.
-
Park JR, Ahn JH, Jung MH, Koh JS, Park W, et al. (2016) Effects of Peroxisome Proliferator-Activated Receptor-d Agonist on Cardiac Healing after Myocardial Infarction. Plos One 11: e0148510.
-
Granger A, Abdullah I, Huebner F, Stout A, Wang T, et al. (2008) Histone deacetylase inhibition reduces myocardial ischemia-reperfusion injury in mice. FASEB J 22: 3549-3560.
-
Zhang LX, DeNicola M, Qin X, Du J, Ma J, et al. (2014) Specific inhibition of HDAC4 in cardiac progenitor cells enhances myocardial repairs. Am J Physiol Cell Physiol 307: C358-372.
-
Svennerholm K, Bergh N, Larsson P, Jern S, Johansson G, et al. (2014) Histone Deacetylase Inhibitor Treatment Increases Coronary t-PA Release in a Porcine Ischemia Model. Plos One 9: e97260.
-
Olesen JB, Hansen PR, Abildstrøm SZ, Andersson C, Weeke P, et al. (2011) Valproate attenuates the risk of myocardial infarction in patients with epilepsy: a nationwide cohort study. Pharmacoepidemiol Drug Saf 20: 146-153.
-
Murakami-Murofushi K, Shioda M, Kaji K, Yoshida S, Murofushi H, et al. (1992) Inhibition of eukaryotic DNA polymerase alpha with a novel lysophosphatidic acid (PHYLPA) isolated from myxoamoebae of Physarumpolycephalum. J Biol Chem 267: 21512-21517.
-
Tsukahara T (2012) The Role of PPARγ in the Transcriptional Control by Agonists and Antagonists. PPAR Res 2012: 362361.
-
Tsukahara T, Tsukahara R, Fujiwara Y, Yue J, Cheng Y, et al. (2010) Phospholipase D2-dependent inhibition of the nuclear hormone receptor PPARgamma by cyclic phosphatidic acid. Mol Cell 39: 421-432.
-
Tsukahara T, Haniu H, Matsuda Y (2014) Cyclic phosphatidic acid inhibits alkyl-glycerophosphate-induced downregulation of histone deacetylase 2 expression and suppresses the inflammatory response in human coronary artery endothelial cells. Int J Med Sci 11: 955-961.
-
Usui T, Okada M, Mizuno W, Oda M, Ide N, et al. (2012) HDAC4 mediates development of hypertension via vascular inflammation in spontaneous hypertensive rats. Am J Physiol Heart Circ Physiol302: H1894-904.
-
Zhang L, Qin X, Zhao Y, Fast L, Zhuang S, et al. (2012) Inhibition of Histone Deacetylases Preserves Myocardial Performance and Prevents Cardiac Remodeling through Stimulation of Endogenous Angiomyogenesis. J Pharmacol & Expe Ther 341: 285-293.
-
Zhao TC, Zhang LX, Cheng G, Liu JT (2010) gp-91 mediates histone deacetylase inhibition-induced cardioprotection. Biochim Biophys Acta 1803: 872-880.
-
Kelly WK, Richon VM, O'Connor O, Curley T, MacGregor-Curtelli B, et al. (2003) Phase I clinical trial of histone deacetylase inhibitor: suberoylanilidehydroxamic acid administered intravenously. Clin Cancer Res 9: 3578-3588.
-
Xie M, Kong Y, Tan W, May H, Battiprolu PK, et al. (2014) Histone deacetylase inhibition blunts ischemia/reperfusion injury by inducing cardiomyocyte autophagy. Circulation 3: 1139-1151.
-
Lopez G, Torres K, Lev D (2011) Autophagy blockade enhances hdac inhibitors' pro-apoptotic effects: Potential implications for the treatment of a therapeutic-resistant malignancy. Autophagy 7: 440-441.
-
Burba I, Colombo GI, Staszewsky LI, De Simone M, Devanna P, et al. (2011) Histone deacetylase inhibition enhances self renewal and cardioprotection by human cord blood-derived CD34 cells. PLoS One 6: e22158.
-
Kong X, Lin Z, Liang D, Fath D, Sang N, et al. (2006) Histone deacetylase inhibitors induce VHL and ubiquitin-independent proteasomal degradation of hypoxia-inducible factor 1alpha. Mol Cell Biol 26: 2019-2028.
-
Nural-Guvener H, Zakharova L, Feehery L, Sljukic S, Gaballa M, et al. (2015) Anti-Fibrotic Effects of Class I HDAC Inhibitor, Mocetinostat Is Associated with IL-6/Stat3 Signaling in Ischemic Heart Failure. Int J Mol Sci 16: 11482-11499.
-
Nural-Guvener HF, Zakharova L, Nimlos J, Popovic S, Mastroeni D, et al. (2014) HDAC class I inhibitor, Mocetinostat, reverses cardiac fibrosis in heart failure and diminishes CD90+ cardiac myofibroblast activation. Fibrogenesis Tissue Repair 7: 1-14.
