

International Journal of Pathology and Clinical Research
JAK2V617F Mutation Affects Downstream LIN28A and HMGA2 Methylation in Myeloproliferative Neoplasms
Chih-Cheng Chen1, Chia-Chen Hsu2, Kuan-Der Lee1, Chia-Chen Chiu2, Hong-Chi Chen3, Tim H.-M. Huang4, Yu-Wei Leu2 and Shu-Huei Hsiao2*
1Department of Hematology and Oncology, Chang Gung Memorial Hospital, Chiayi, Chang Gung University College of Medicine, Taiwan; Chang Gung Institute of Technology, Taoyuan, Taiwan
2Human Epigenomics Center, Department of Life Science, Institute of Molecular Biology and Institute of Biomedical Science, National Chung Cheng University, Chiayi, Taiwan
3Department of Life Science and Gene Therapy Division, Tzu-Chi University and Hospital, Hualien, Taiwan
4Cancer Therapy and Research Center, Department of Molecular Medicine and Institute of Biotechnology, School of Medicine, The University of Texas Health Science Center at San Antonio, San Antonio, TX, 78229, USA
*Corresponding author:
Shu-Huei Hsiao, Human Epigenomics Center, Department of Life Science and Institute of Molecular Biology, National Chung Cheng University, 168, University Rd., Min-Hsiung, Chiayi, Taiwan, 62102, Tel: 886-5-272-0411, 53202, Fax: 886-5-272-2871, E-mail: bioshh@ccu.edu.tw
Int J Pathol Clin Res, IJPCR-2-027, (Volume 2, Issue 1), Mini Review; ISSN: 2469-5807
Received: January 08, 2016 | Accepted: February 26, 2016 | Published: February 29, 2016
Citation: Chih-Cheng C, Chia-Chen H, Kuan-Der L, Chia-Chen C, Hong-Chi C, et al. (2016) JAK2V617F Mutation Affects Downstream LIN28A and HMGA2 Methylation in Myeloproliferative Neoplasms. Int J Pathol Clin Res 2:027. 10.23937/2469-5807/1510027
Copyright: © 2016 Chih-Cheng C, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Myeloproliferative neoplasm (MPN) is a hematological disease. The Janus kinase 2 (JAK2) V617F mutation (valine → phenylalanine at amino acid 617) has been identified in fifty (essential thrombocythemia) to ninety (polycythemia vera) percent of patients. JAK2 governs the phosphorylation of signal transducer and activator of transcription 3 (STAT3), a transcription factor. Through this signaling relay, we hypothesized that JAK2V617F expression results in specific epigenetic modifications, including DNA methylation, and we aimed to identify these modifications. Methylation-specific PCR (MSP) was used to detect changes within JAK2-targeted genes before and after JAK2V617F expression and in MPN patient samples. To identify JAK2 targets, whole-genome sequencing was used to measure the enrichment of phosphorylated STAT3 (pSTAT3), enhancer of zeste homolog (EZH2), and methylated DNA before and after JAK2V617F expression. We then tested whether methylation was altered concurrently. Methylation of JAK2-regulated genes Lin28A and HMGA2 is affected by JAK2V617F in BA/F3 cells; similar changes in methylation were also observed in MPN samples expressing JAK2V617F. JAK2V617F also affected pSTAT3 and EZH2 binding. The methylation profile was specifically altered. Therefore, JAK2V617F affected both the DNA methylation profile and binding of pSTAT3 and EZH2 at specific promoter sites. Specific changes in EZH2 binding and methylation suggest that aberrant JAK2 signaling results in the accumulation of heritable changes. Methylation changes in patient samples expressing JAK2V617F suggest that these changes occur in vivo. Future study of affected epigenomes is necessary to identify the critical factors required for cellular transformation.
Keyword
DNA methylation, Myeloproliferative neoplasms, Janus kinase 2 (JAK2), Epigenetics
Introduction
Myeloproliferative neoplasms (MPNs) are hematological neoplasms that arise from bone marrow and can progress to myelodysplastic syndrome and acute myeloid leukemia [1]. Next-generation genomic and exomic sequencing has revealed that the Janus kinase 2 (JAK2, NM_004972) V617F mutation (valine → phenylalanine at amino acid 617) is a major contributor to MPN transformation [2,3]. Clinical and animal models have shown that the JAK2V617F mutation is sufficient for transformation [4]. Other studies have indicated that JAK2V617F contributes to cellular transformation by either interfering with the phosphorylation of signal transducer and activator of transcription (STAT) proteins or altering tet methylcytosine dioxygenase 2 (TET2, NM_017628) [5-7].
The lineage/clonal selection theory for cellular transformation may help identify the effector genes whose functions are crucial for MPN tumorigenesis [8]. MPNs originate from hematopoietic stem cells carrying compartmental mutations such as JAK2V617F [9]. JAK2 signaling regulates target gene expression by altering the phosphorylation and nuclear localization of STATs [5,10]. To provide selectable gene expression that promotes transformation of certain cellular lineages, specific signals relayed to the cell nucleus could be memorized as heritable codes such as epigenetic modifications [11,12]. Because expression of TET2, a demethylation enzyme, was found to be inhibited in MPNs [13,14], the related epigenome, especially the methylome, might then be abnormal in MPNs [7]. Abnormal cell signaling may alter the methylation of genes involved in cell fate, which would change the expression of these critical genes and their contribution to the transformation process.
Signaling-specific epigenetic changes were identified in cultured cells and may occur in vivo [12]. For example, nuclear receptors such as the estrogen receptor (ER) are transcription factors that bind directly to their target promoters and change their methylation states [11,15]. Previous data indicate that silencing upstream ER signals suppressed ER-targeted loci and allowed for DNA methylation [11]. These epigenetic changes are regulated by the ER and associated co-regulators Myc and STAT [11,16,17]. New techniques have made tracking these reproducible modifications possible, even when the systems are regulated by multiple factors in vivo; consequently, signaling-specific abnormal DNA methylation can now be assessed in tumor samples [18,19]. In addition, similar signaling-specific epigenomic searches may reduce the number of potential targets that typically accumulate in “big data” genomics and epigenomics studies [20]. Furthermore, the correlation of epigenomic changes identified in target loci and in vivo samples is important for therapeutic applications [12].
In this study, JAK2V617F was overexpressed in BA/F3 (BaF3) cells as described by Pradhan et al. [21], and the methylation of JAK2-targeted loci, such as Lin28A [22], HMGA2 [23], and enhancer of zeste homolog (EZH2) [24] was assessed to determine whether JAK2V617F expression results in specific epigenetic modifications, including DNA methylation.
Materials and Methods
Cell culture
BaF3 cells were cultured in RPMI 1640 media supplemented with 10% fetal bovine serum (Invitrogen), 100 mg/ml penicillin/streptomycin (Invitrogen), 10 ng/ml human recombinant interleukin-3 (R&D), and 2 mM L-glutamine (Invitrogen). Cells were incubated in 5% CO2 at 37°C.
Chromatin immunoprecipitation sequencing (ChIP-seq)
DNA in control or JAK2V617F-transfected BaF3 [21] cells (1 × 108) was cross-linked by the addition of 1% formaldehyde for 10 minutes. Cross-linking was stopped by the addition of 0.125 M glycine. Cells were washed twice in phosphate-buffered saline (PBS) and lysed using cell lysis buffer (5 mM PIPES, pH 8.0, 85 mM KCl, and 0.5% NP-40). Lysates were passed through a Dounce tissue homogenizer 20 times, and the nuclei were isolated by centrifugation (3000 × g, 15 minutes). DNA was released from isolated nuclei using ChIP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-Cl, pH 8.1, and 167 mM NaCl) and sonicated into fragments (size ~200 base pairs). Antibodies against pSTAT3 (Millipore, 05-485) and EZH2 (Millipore, 07-689) were added to the fragmented DNA. After an overnight incubation, 60 μl Protein A/agarose were added to the samples for 4 hours. Samples were washed with a low-salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1, and 150 mM NaCl), a high-salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1, and 500 mM NaCl), a LiCl buffer (0.25 M LiCl, 1% IGEPAL-CA630, 1% deoxycholate, 1 mM EDTA, and 10 mM Tris-HCl, pH 8.1), and TE (100 mM Tris-HCl, pH 8.0, and 1 mM EDTA). The DNA was then eluted in buffer (1% SDS and 0.1 M sodium bicarbonate). Samples were incubated in 5 M NaCl at 65°C to reverse the cross-linking, and proteins were digested using proteinase K. After phenol-chloroform extraction, DNA was precipitated with twice volumes of alcohol, quantified, and sequenced (Welgene, Taiwan).
Methylation-binding protein/domain capture sequencing (MBD-seq)
Methylated DNA from control and JAK2V617-transfected BaF3 cells was isolated using the MethylMiner Methylated DNA Enrichment kit (Invitrogen) following the manufacturer's instructions. DNA was sheered into approximate 200-base-pair fragments, quantified, and sequenced (Welgene, Taiwan).
Semi-quantitative real-time methylation-specific PCR (qMSP)
We performed qMSP experiments and quantified the PCR products as described in Yan et al. [25]. Bisulfite-converted genomic DNA (0.5 μg initially) was used to perform real-time PCR with methylation-specific primers (Table S1). The reactions were performed using the SYBR Green I PCR kit (Roche) and an iQ5 Real-Time PCR instrument (Bio-Rad). Melting curves were assessed to ensure amplification of specific amplicons. Serial dilutions of Col2A1 (NM_033150)-amplified, bisulfite-converted, universal methylated DNA (Millipore) were used to generate a standard curve. The percentage of methylation was calculated as: [Mean of target gene] × 100%/[Mean of Col2A1].
Statistical analyses
Paired Student's t test was used to examine the methylation differences between control and JAK2V617F-overexpressing BaF3 cells. The sequence data were quality controlled and aligned. Peaks were identified using the CLC Genomics Workbench package (CLC bio). Spearman's coefficients were deduced from sorted methylation differences from either wild type or JAK2V617F MPNs.
Results
JAK2V617F overexpression results in methylation differences at specific loci
To test whether JAK2V617F is sufficient to alter the methylation profile, the methylation of JAK2-targeted loci [Lin28A (human, NM_024674; mouse, NM_145833), HMGA2 (human, NM_003483; mouse, NM_010441), and EZH2 (human, NM_003456; mouse, NM_007971)] was examined in BaF3 cells expressing JAK2V617F. L1TD1 was used as a negative control in vivo (Figure 1a and Figure 1b). We found significant methylation differences within the Lin28A and HMGA2 promoters when control samples were compared to cells expressing JAK2V617F (Figure 1a). Therefore, JAK2V617F might be sufficient to change the methylation of JAK2-targeted loci. In patient samples, the methylation profiles within these two loci also varied (Figure 1c and Figure 1d), which suggested changes to the epigenome in transformed cells; however, the role of JAK2V617F to the altered epigenetic landscape was unclear (Figure 1b, Figure 1c, Figure 1d and Figure S1, patient information in Table S2).
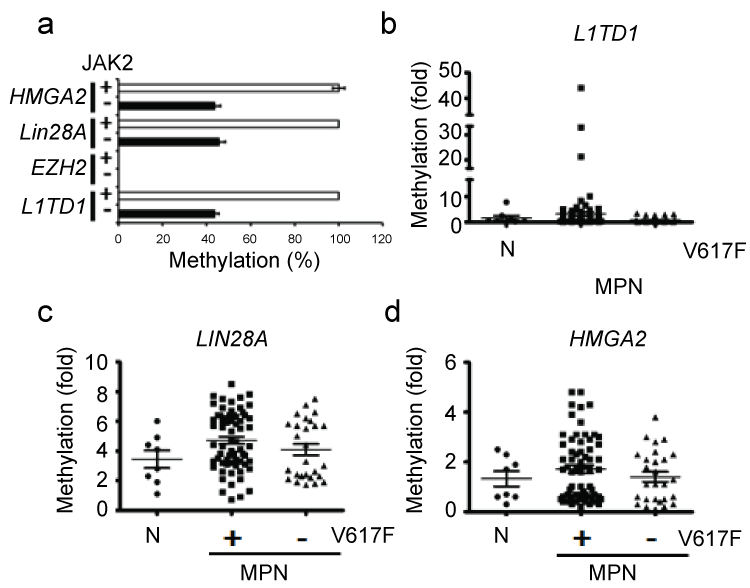
.
Figure 1: JAK2V617F-associated methylation changes in BaF3 cells and MPN samples. (a) qMSP was used to measure methylation within the HMGA2, Lin28A, EZH2, and L1TD1 promoters in control BaF3 cells (JAK2, +) and BaF3 cells expressing JAK2V617F (JAK2, -). In human normal control (N) and MPN samples with (V617F, +) or without the JAK2V617F mutation (V617F, -), qMSP was used to detect methylation within the (b) L1TD1, (c) Lin28A, and (d) HMGA2 promoters.
View Figure 1
Characterization of the affected epigenome in JAK2V617F-expressing cells
JAK2 governs the phosphorylation of STATs, such as STAT3; pSTAT3 enters the nucleus to regulate the expression of select target genes. If changes in the expression of these targeted loci were a result of epigenetic changes, such as DNA methylation, then these changes could be caused by aberrant JAK2 signaling. This finding would suggest that altered JAK2 signaling and heritable JAK2-dependent epigenetic changes are both associated with tumorigenesis and that JAK2 oncogenic effector genes are modified epigenetically. Therefore, identifying the epigenetic changes associated with the JAK2V617F mutation is one way to find important oncogenic genes.
To test this idea, ChIP-seq and MBD-seq were performed to identify alterations in pSTAT3 and EZH2 binding, and alterations in the methylation profiles, respectively. EZH2 is a polycomb group (PcG) protein that represses associated genes [26,27]. Loss of PcG proteins and their substrates induce stem cell differentiation [28-30]; the same PcG-regulated loci and epigenetic modifications have been observed in cancer stem-like cells [31]. Therefore, the detected changes in EZH2 binding might indicate an altered cell fate [26,27]. Using the same rationale, we assessed DNA methylation in JAK2V617F-expressing cells, because changes in DNA methylation are associated with cell fate alterations [12]. We observe that pSTAT3 targets differed in cells expressing the JAK2V617F mutation (green → red bars, Figure 2). Changes in DNA-associated EZH2 (blue → orange bars, Figure 2) and DNA methylation (grey → black bars, Figure 2) were also revealed. Because our experiment assessed global alterations, we also identified loci unaltered by the JAK2V617F mutation, which could serve as negative controls. The candidate targets are listed in Table S3.
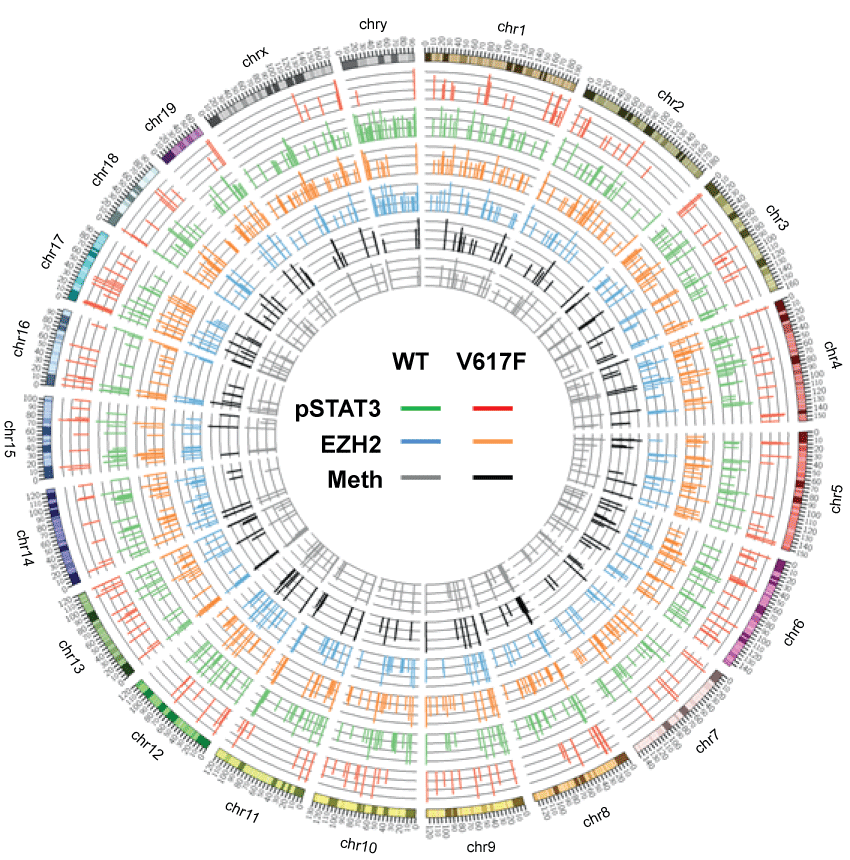
.
Figure 2: Circos plot of pSTAT3 binding, EZH2 binding, and DNA methylation in BaF3 cells with and without JAK2V617F. After ChIP-seq and MBD-seq, the position and log-transformed reads of pSTAT3 (outer two circles, red and green bars) and EZH2 (middle two circles, orange and blue bars) enrichment, and DNA methylation (inner circles, black and grey bars) were aligned according to their relative cytological positions. The chromosomes are listed in the outermost ring.
View Figure 2
Subtle epigenetic changes in JAK2V617F-expressing cells
JAK2 downstream targets were affected differently, revealing the specificity and complexity of the epigenetic alternations caused by JAK2V617F. These complexities were revealed by examining the binding differences within the top JAK2V617F-altered pSTAT3-enriched genes in Table S3. Only five percent of wild type pSTAT3 targets still can be recognized in JAK2V617F cells. In Figure 3 (Top), Sfi1 (NM_030207) is used as control locus that its pSTAT3 enrichment is not affected by the JAK2V617F mutation. pSTAT3 enrichment within Litaf (NM_019980) was reduced in JAK2V617F mutation. For the Lars2 (NM_153168) and Aida (NM_181732) promoter, pSTAT3 was significantly enriched in JAK2V617-expressing cells but not in the wild type cells. These changes also demonstrate the specificity of the sequencing results.
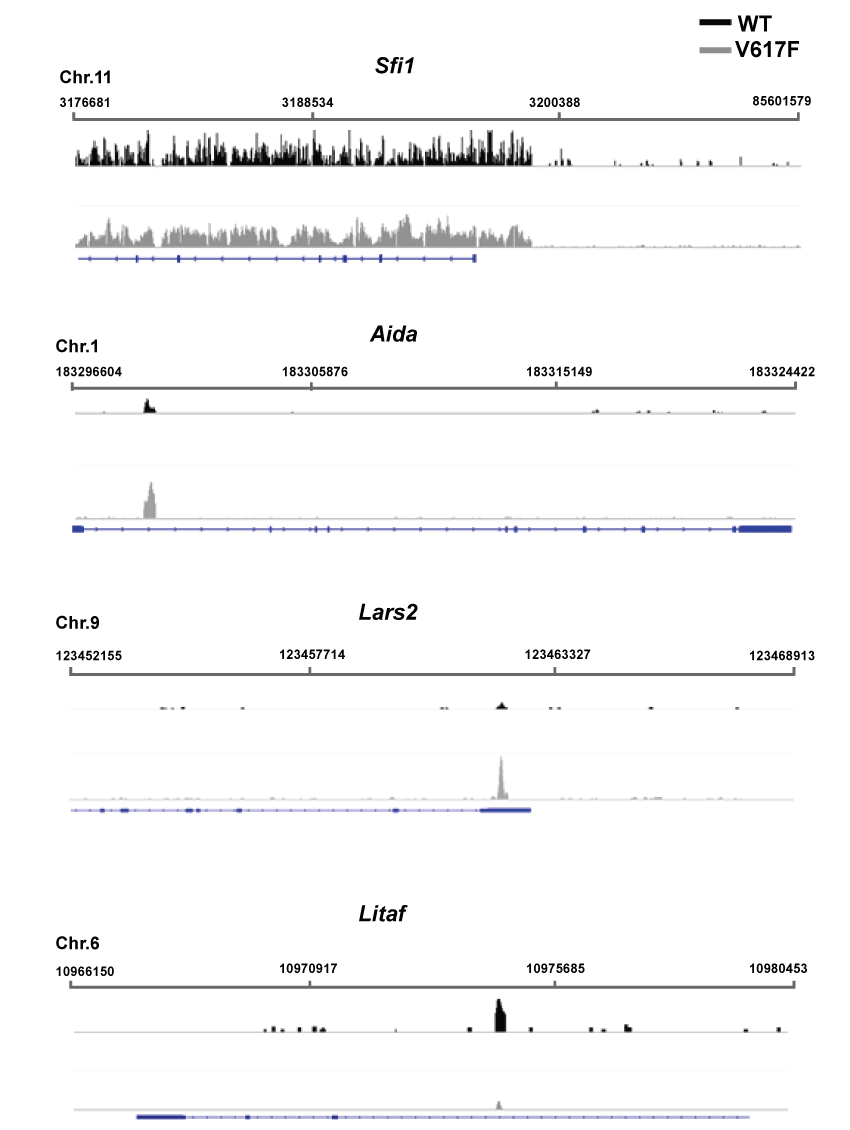
.
Figure 3: Examples of pSTAT3 enrichment in different loci before and after JAK2V617F expression in BaF3 cells. The log-transformed pSTAT3 enrichment in wild type (WT, black) and JAK2V617F-expressing (V617F, grey) BaF3 cells is plotted under the designated genes. The upper bars and numbers represent the chromosomal region analyzed. The blue lines represent the entire gene, whereas the boxes represent the exons.
View Figure 3
Associated methylation changes between Lin28A and HMGA2 in MPN samples
Lin28A and HMGA2 are regulated by JAK2 signaling [22,23]; the associated methylation changes in these genes in vivo would indicate the existence of a causal relationship in patient samples. In Figure 4a, methylation heat maps were generated when data were sorted according to L1TD1 (a non-JAK2 regulated gene, Figure 4a, left panel) or Lin28A (Figure 4a, right panel) methylation. Similar heat map patterns were observed for Lin28A and HMGA2; however, they differed from that for L1TD1. The methylation changes between HMGA2 and Lin28A showed a significant association (Figure 4b). The associations between Lin28A and L1TD1 (Figure 4c) and between HMGA2 and L1TD1 (Figure 4d) are not significant, compared to the association observed between Lin28A and HMGA2. In contrast to the concurrent alterations in Lin28A and HMGA2 methylation, the methylation difference of the three genes in the MPN samples between with or without the JAK2V617F mutation are all not significant, indicating that other factors interfere with the establishment of downstream abnormal epigenetic modifications.
Discussion
Hematopoietic differentiation is a well-characterized process; JAK2 signaling is known to control normal differentiation [32]. The JAK2V617F mutation has been detected in hematopoietic illnesses such as MPN, suggesting that JAK2 is responsible for normal differentiation and that alternations in gene expression downstream of JAK2 signaling might be involved in the transformation process [2,4]. Presently, numerous candidate genes have been identified from whole-genome association and methylome studies. Signaling-based searches have recently gained attention to help narrow the list of candidate genes and ultimately identify those involved in transformation [33,34]. The rationale that aberrant signaling leads to specific epigenetic changes that are inherited by somatic cells during cellular transformation has bolstered the viability of using such studies [12]. Because the effect of JAK2 signaling on the development of MPN from hematopoietic precursors is well known, the use of the JAK2V617F mutant in future studies might accelerate the discovery of important mediators of this transformation.
The complexity of altered pSTAT3 binding in JAK2V617F-expressing cells demonstrates the specificity of the sequencing results and reveals that a more complex regulatory system is involved in the development of MPN (Figure 2 and Figure 3). If the JAK2-initiated, pSTAT3-determined, epigenetic changes were regulated through a simple, single molecular machine, then we would expect the JAK2V617F-induced pSTAT3 association to be the same for pSTAT3-binding loci. However, we observed locus-specific changes, and the loci were difficult to organize into specific patterns (Figure 3). There are likely more regulators, working in trans or in cis, involved in MPN development, and identifying these regulators will be the topic of future studies. The identification of true targets might also reveal additional complexity, because it is unlikely that all of the regulators are involved in transformation. Also, the switched EZH2 and DNA methylation pattern (Figure 2 and Figure 3) after JAK2V617F-expression revealed the changed inheritable epigenomic marks and potentially the changed cell fates. EZH2 belongs to the Polycomb group proteins and is responsible for the maintenance of stemness in several progenitor cells [35]. JAK2V617F-expression is one of the mutations that was reported to cause the transformation from hematopoietic stem cell niche [10,36]; and there are more accumulated, ordered mutations that might lead to different types of MPNs [37]. Tracing specific mutations-induced epigenetic changes might help the reduction of target findings in MPNs.
Whether the above-mentioned signal/mutation generated epigenetic changes occurred during the in vivo transformation remains to be validated. Lin28A and HMGA2 are reported to be associated with JAK2 mutated MPNs [38-41]. Lin28A itself is a stem cell mark [42] that regulates the let7 microRNAs formation [43]. Let 7 miRNAs also regulate the carcinogenesis and stem cell phenotype through its regulation on HMGA2 [44]. HMGA2 is able to regulate the telomere stability and proliferation [45] in cells and is related to cellular transformation. Associated methylation changes of both genes (Figure 4) revealed the signal/mutation specific epigenetic changes occurred in vivo. Together, if more mutation-specific downstream epigenetic changes can be identified, more different downstream target genes for different types of MPNs could be identified and might be used for their diagnosis or designs of treatments.
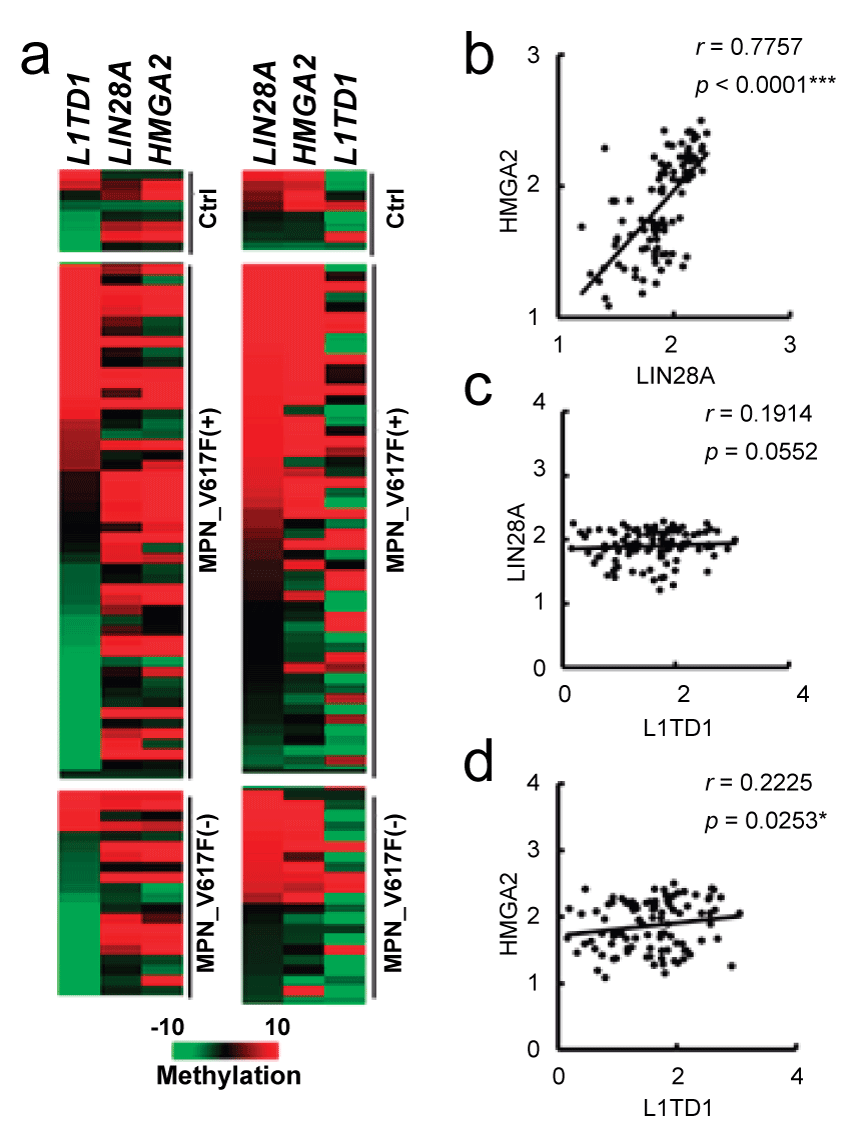
.
Figure 4: Concurrent methylation changes within Lin28A and HMGA2 in MPN samples. (a) DNA methylation heat maps of L1TD1, Lin28A, and HMGA2. Left, assorted heat map according to the methylation state in L1TD1. Right, assorted heat map according to the methylation state in Lin28A. Ctrl: normal blood; MPN_V617(+): MPN samples with the JAK2V617F mutation; MPN_V617(-): MPN samples without the JAK2V617F mutation. Association analyses between the methylation states: (b) Lin28A and L1TD1; (c) HMGA2 and Lin28A; and (d) HMGA2 and L1TD1.
View Figure 4
Human subjects
MPN samples were collected from Chang Gung Memorial Hospital, Chiayi, Taiwan in accordance with Institutional Review Board regulations (IRB100-1520B, 102-5970B). The pathological characteristics of the patient samples are listed in Table S2.
Author contributions
The first two authors contributed equally.
Acknowledgements
CCC and YWL are supported by Chang Gung Memorial Hospital (NMRPD1D0982, CORPG6B0373 and CMRPG6F0091). YWL and KDL are supported by MoST 103-2314-B-182A-090. YWL and SHH are supported by MoST (NSC 102-2320-B-194-003-MY3, MoST 104-2320-B-194-001) and NHRI (NHRI-EX102-10259NI).
References
-
Silvennoinen O, Hubbard SR (2015) Molecular insights into regulation of JAK2 in myeloproliferative neoplasms. Blood 125: 3388-3392.
-
Milosevic Feenstra JD, Nivarthi H, Gisslinger H, Leroy E, Rumi E, et al. (2015) Whole exome sequencing identifies novel MPL and JAK2 mutations in triple negative myeloproliferative neoplasms. Blood 127: 325-332.
-
Kirschner MM, Schemionek M, Schubert C, Chatain N, Sontag S, et al. (2015) Dissecting Genomic Aberrations in Myeloproliferative Neoplasms by Multiplex-PCR and Next Generation Sequencing. PLoS One 10: e0123476.
-
Xing S, Wanting TH, Zhao W, Ma J, Wang S, et al. (2008) Transgenic expression of JAK2V617F causes myeloproliferative disorders in mice. Blood 111: 5109-5117.
-
Oh ST, Simonds EF, Jones C, Hale MB, Goltsev Y, et al. (2010) Novel mutations in the inhibitory adaptor protein LNK drive JAK-STAT signaling in patients with myeloproliferative neoplasms. Blood 116: 988-992.
-
de Freitas RM, da Costa Maranduba CM (2015) Myeloproliferative neoplasms and the JAK/STAT signaling pathway: an overview. Rev Bras Hematol Hemoter 37: 348-353.
-
Ko M, Huang Y, Jankowska AM, Pape UJ, Tahiliani M, et al. (2010) Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature 468: 839-843.
-
Fisher DAC (2012) Clonal Evolution Revealed by Whole Genome Sequencing in a Case of Primary Myelofibrosis Transformed to Secondary Acute Myeloid Leukemia. Blood 120.
-
Lundberg P, Takizawa H, Kubovcakova L, Guo G, Hao-Shen H, et al. (2014) Myeloproliferative neoplasms can be initiated from a single hematopoietic stem cell expressing JAK2-V617F. J Exp Med 211: 2213-2230.
-
Vainchenker W, Constantinescu SN (2013) JAK/STAT signaling in hematological malignancies. Oncogene 32: 2601-2613.
-
Leu YW, Yan PS, Fan M, Jin VX, Liu JC, et al. (2004) Loss of estrogen receptor signaling triggers epigenetic silencing of downstream targets in breast cancer. Cancer Res 64: 8184-8192.
-
Hsiao SH, Huang TH, Leu YW (2009) Excavating relics of DNA methylation changes during the development of neoplasia. Semin Cancer Biol 19: 198-208.
-
Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, et al. (2011) Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 20: 11-24.
-
Tefferi A, Lim KH, Levine R (2009) Mutation in TET2 in myeloid cancers. N Engl J Med 361: 1117.
-
Shi H, Wei SH, Leu YW, Rahmatpanah F, Liu JC, et al. (2003) Triple analysis of the cancer epigenome: an integrated microarray system for assessing gene expression, DNA methylation, and histone acetylation. Cancer Res 63: 2164-2171.
-
Jhanwar-Uniyal M (2003) BRCA in cancer, cell cycle and genomic stability. Front Biosci 8: s1107-1117.
-
Cheng AS, Jin VX, Fan M, Smith LT, Liyanarachchi S, et al. (2006) Combinatorial analysis of transcription factor partners reveals recruitment of c-MYC to estrogen receptor-alpha responsive promoters. Molecular Cell 21: 393-404.
-
Ray D, Kwon SY, Ptasinska A, Bonifer C (2013) Chronic growth factor receptor signaling and lineage inappropriate gene expression in AML: the polycomb connection. Cell Cycle 12: 2159-2160.
-
Halvorsen AR, Helland A, Fleischer T, Haug KM, Grenaker Alnaes GI, et al. (2014) Differential DNA methylation analysis of breast cancer reveals the impact of immune signaling in radiation therapy. Int J Cancer 135: 2085-2095.
-
Maze I, Shen L, Zhang B, Garcia BA, Shao N, et al. (2014) Analytical tools and current challenges in the modern era of neuroepigenomics. Nat Neurosci 17: 1476-1490.
-
Pradhan A, Lambert QT, Reuther GW (2007) Transformation of hematopoietic cells and activation of JAK2-V617F by IL-27R, a component of a heterodimeric type I cytokine receptor. Proc Natl Acad Sci U S A 104: 18502-18507.
-
Ma W, Kantarjian H, Zhang X, Wang X, Zhang Z, et al. (2010) JAK2 exon 14 deletion in patients with chronic myeloproliferative neoplasms. PLoS One 5: e12165.
-
Grosso S, Puissant A, Dufies M, Colosetti P, Jacquel A, et al. (2009) Gene expression profiling of imatinib and PD166326-resistant CML cell lines identifies Fyn as a gene associated with resistance to BCR-ABL inhibitors. Mol Cancer Ther 8: 1924-1933.
-
Sarkar D, Leung EY, Baguley BC, Finlay GJ, Askarian-Amiri ME (2015) Epigenetic regulation in human melanoma: past and future. Epigenetics 10: 103-121.
-
Yan PS, Venkataramu C, Ibrahim A, Liu JC, Shen RZ, et al. (2006) Mapping geographic zones of cancer risk with epigenetic biomarkers in normal breast tissue. Clin Cancer Res 12: 6626-6636.
-
Béguelin W, Popovic R, Teater M, Jiang Y, Bunting KL, et al. (2013) EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell 23: 677-692.
-
Chou RH, Yu YL, Hung MC (2011) The roles of EZH2 in cell lineage commitment. Am J Transl Res 3: 243-250.
-
Agger K, Cloos PA, Christensen J, Pasini D, Rose S, et al. (2007) UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature 449: 731-734.
-
Jepsen K, Solum D, Zhou T, McEvilly RJ, Kim HJ, et al. (2007) SMRT-mediated repression of an H3K27 demethylase in progression from neural stem cell to neuron. Nature 450: 415-419.
-
Lan F, Bayliss PE, Rinn JL, Whetstine JR, Wang JK, et al. (2007) A histone H3 lysine 27 demethylase regulates animal posterior development. Nature 449: 689-694.
-
Ohm JE, McGarvey KM, Yu X, Cheng L, Schuebel KE, et al. (2007) A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet 39: 237-242.
-
Ward AC, Touw I, Yoshimura A (2000) The Jak-Stat pathway in normal and perturbed hematopoiesis. Blood 95: 19-29.
-
Jost E, do O N, Dahl E, Maintz CE, Jousten P, et al. (2007) Epigenetic alterations complement mutation of JAK2 tyrosine kinase in patients with BCR/ABL-negative myeloproliferative disorders. Leukemia 21: 505-510.
-
Lan X, Adams C, Landers M, Dudas M, Krissinger D, et al. (2011) High resolution detection and analysis of CpG dinucleotides methylation using MBD-Seq technology. PLoS One 6: e22226.
-
Wang L, Brown JL, Cao R, Zhang Y, Kassis JA, et al. (2004) Hierarchical recruitment of polycomb group silencing complexes. Mol Cell 14: 637-646.
-
Arranz L, Sánchez-Aguilera A, Martín-Pérez D, Isern J, Langa X, et al. (2014) Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature 512: 78-81.
-
Ortmann CA, Kent DG, Nangalia J, Silber Y, Wedge DC, et al. (2015) Effect of mutation order on myeloproliferative neoplasms. N Engl J Med 372: 601-612.
-
Harada-Shirado K, Ikeda K, Ogawa K, Ohkawara H, Kimura H, et al. (2015) Dysregulation of the MIRLET7/HMGA2 axis with methylation of the CDKN2A promoter in myeloproliferative neoplasms. Br J Haematol 168: 338-349.
-
Ikeda K, Mason PJ, Bessler M (2011) 3'UTR-truncated Hmga2 cDNA causes MPN-like hematopoiesis by conferring a clonal growth advantage at the level of HSC in mice. Blood 117: 5860-5869.
-
Ikeda K, Ogawa K, Takeishi Y (2012) The role of HMGA2 in the proliferation and expansion of a hematopoietic cell in myeloproliferative neoplasms. Fukushima J Med Sci 58: 91-100.
-
Saeidi K (2016) Myeloproliferative neoplasms: Current molecular biology and genetics. Crit Rev Oncol Hematol 98: 375-389.
-
Carmel-Gross I, Bollag N, Armon L, Urbach A (2016) LIN28: A Stem Cell Factor with a Key Role in Pediatric Tumor Formation. Stem Cells Dev .
-
Heo I, Joo C, Cho J, Ha M, Han J, et al. (2008) Lin28 mediates the terminal uridylation of let-7 precursor MicroRNA. Mol Cell 32: 276-284.
-
Madison BB, Jeganathan AN, Mizuno R, Winslow MM, Castells A5, et al. (2015) Let-7 Represses Carcinogenesis and a Stem Cell Phenotype in the Intestine via Regulation of Hmga2. PLoS Genet 11: e1005408.
-
Shi Z, Li X, Wu D, Tang R, Chen R, et al. (2015) Silencing of HMGA2 suppresses cellular proliferation, migration, invasion, and epithelial-mesenchymal transition in bladder cancer. Tumour Biol .
