

International Journal of Pathology and Clinical Research
Intracellular Effectors of Synaptic Dysfunction and Neuroinflammation in Alzheimer's and other Neurodegenerative Diseases
Zen Kouchi*
Department of Pathology, Institute for Developmental Research, Aichi Human Service Center, Japan
*Corresponding author:
Zen Kouchi, Department of Pathology, Institute for Developmental Research, Aichi Human Service Center, 713-8 Kamiya-cho, Kasugai-city, Aichi 480-0392, Japan, Tel: +81 568 88 0811, Fax: +81 568 88 0829, E-mail: zkouchi@inst-hsc.jp
Int J Pathol Clin Res, IJPCR-2-021, (Volume 2, Issue 1), Review Article; ISSN: 2469-5807
Received: November 26, 2015 | Accepted: January 26, 2016 | Published: February 01, 2016
Citation: Kouchi Z (2016) Intracellular Effectors of Synaptic Dysfunction and Neuroinflammation in Alzheimer's and other Neurodegenerative Diseases. Int J Pathol Clin Res 2:021. 10.23937/2469-5807/1510021
Copyright: © 2016 Kouchi Z. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Synaptic function is maintained by dynamic processes governed by regulators of plasticity or morphogenesis of pre- and postsynaptic compartments. Synaptic dysfunction often precedes neuronal death around the time of disease onset in neurodegenerative diseases, including Alzheimer's disease (AD), and dysregulated microglia cause prolonged neuroinflammation with severe clinical symptoms. Although impaired amyloid beta (Aβ) clearance by astrocyte-derived apolipoprotein E4 (ApoE4) has been considered to be the main contributor of sporadic AD, intracellular effectors, such as cell-adhesion regulatory proteins or lipophilic mediators, have been shown to regulate synaptic homeostasis, and are further involved in regulating chronic propagation of inflammation during the neurodegenerative process including AD. Catenin family proteins, such as β-catenin and p120 catenin, regulate cadherin trafficking and cytoskeletal rearrangement. Aberrant catenin signaling has been shown to play a role in the neuronal dysfunction seen in AD or Parkinson disease (PD) models with abnormal processing of amyloid precursor protein (APP) or oxidative vulnerability. The most abundant lipophilic endocannabinoid (eCB) in the brain, 2-arachidonoylglycerol (2-AG), is primarily generated by sequential hydrolysis of phosphatidylinositol-4,5-bisphosphate (PIP2) by phospholipase C and diacylglycerol lipases, both important for normal synaptic transmission. Imbalance of PIP2 metabolism is implicated in the signaling of presenilin 1 (PS1) mutations causing familial AD or oligomeric amyloid beta (Aβ) peptide administration. Furthermore, monoacylglycerol lipase (MAGL) degrades 2-AG and has been known to terminate cannabinoid receptor (CBR)-mediated signaling; additionally MAGL plays a proinflammatory role in progression of neurodegeneration independent of CBR signaling. Here, we focus on several catenins regulating cadherin-mediated signaling, and lipid signal modulators organizing phosphoinositide (PI) or 2-AG metabolism in neurons and glia; special attention is given to how the microglial surveillance system is disorganized during the progression of neurodegeneration in AD and PD models.
Keyword
Alzheimer's disease, 2-Arachidonoylglycerol, Cadherin, Catenin, Microglia, Monoacylglycerol Lipase, Neuroinflammation, Presenilin
Introduction
Synaptic morphogenesis and plasticity are regulated by neuronal activity to form efficient neuronal networks based on somatosensory inputs or behavioral experiences. Enhanced neuronal activity triggers reorganization of the actin cytoskeleton in presynaptic compartments and induces coordinated changes in apposed postsynaptic density with actin dynamics [1]. Numerous cell-adhesion molecules are involved in these processes, and classical cadherins are most characterized and important for synaptic formation or dendritic arborization [2,3]. Several modulators of neurotransmission, besides cell-adhesion proteins, play a role in synaptic plasticity. Neuronal activity-dependent processing of amyloid precursor protein (APP) is required for synaptic homeostasis, but excess production of amyloid β (Aβ) can depress synaptic transmission, resulting in cognitive decline, as seen in early pathogenesis of AD [4]. Synaptic dysfunction has been shown to precede Aβ deposition and neurofibrillary tangle formation in AD model mice harboring knockin mutant presenilin 1 (PS1M146V), mutant tau protein (tauP301L), and Swedish APP (APPSwe), which simulate the regional impact of Aβ plaques and neurofibrillary tangles observed in AD pathology [5]. Underlying regulatory mechanism of APP processing by PS1 interacting proteins, such as catenin, and that of inflammatory balance in dopaminergic neurons, have also been elucidated, and catenin family proteins play an important role in AD and PD pathogenesis with different mechanisms [6-12]. Catenins including p120, β- or δ-catenin also play an important role in actin reorganization through Rho family proteins at dendritic spines and synapses, whilst genetic inactivation can cause severe synaptic dysfunction (Table 1) [13-16]. Furthermore, δ-catenin, a neuron-specific catenin involved in dendritic branching, is recognized as a causative gene of Cri-du-chat syndrome, which presents severe cognitive impairment and mental retardation [17].
In addition to these regulatory proteins localized at the synaptic compartments, several membrane-derived lipid metabolites are important for the regulation of synaptic transmission. Phosphatidylinositol-4,5-bisphosphate (PIP2) is critical for exocytosis of synaptic vesicles and membrane invagination for recycling processes, with several regulators including synaptotagmin and small GTPases [18,19]. Following depolarization-induced Ca2+ influx or activation of metabotropic receptors, PIP2 is hydrolyzed by phospholipase C and generated diacylglycerol (DAG) is subsequently degraded by sn-1-specific diacylglycerol lipases (DAGLα and DAGLβ). The most abundant endocannabinoid (eCB), 2-arachidonoylglycerol (2-AG) is generated by DAGL-mediated hydrolysis at postsynaptic compartments, traversing synaptic clefts and repressing neurotransmitter release [20,21]. 2-AG production is referred to as “on demand” biogenesis with neuronal activity, whilst most 2-AG is removed from the synaptic cleft and hydrolyzed by monoacylglycerol lipase (MAGL) at presynaptic compartments [22]. 2-AG is known to act as a ligand for cannabinoid receptors (CBRs); CB1R is predominantly expressed in neurons, whereas CB2R mainly resides in immune cells. The neuronal MAGL-dependent regulation of presynaptic CB1R signaling is known as depolarization-induced suppression of excitation (DSE) and inhibition (DSI) in excitatory and inhibitory neurons, respectively [23,24], whereas involvement of MAGL in synaptic dysfunction and Aβ deposition has also been reported in AD mouse models [25-27].
A similar 2-AG generating mechanism catalyzed by PLC and DAGLs is preserved in microglia expressing purinergic receptors that respond to extracellular ATP [24]. Noticeably, MAGL-mediated production of proinflammatory arachidonic acid (AA) is required for microglial activation and generation of inflammatory cytokines. Microglial MAGL expression is known to be upregulated in inflammatory conditions, such as Aβ accumulation or LPS treatment, and MAGL is involved in phagocytic activity of activated microglia [28,29]. In immune cells CB2R is hardly detectable under normal conditions but induced upon neuronal inflammation, as 2-AG activates CB receptor (CBR) signaling, resulting in microglial migration [30]. Since modulation of CB1R signaling induces severe psychotropic effects, development of CB2R-specific chemicals has been sought after for the resolution of neuroinflammation as a clinical issue.
As for other lipid regulators, low-density lipoprotein receptor (LDLR) family such as low-density lipoprotein related proteins (LRPs) regulates internalization of apolipoprotein E (ApoE)-containing lipoprotein at synapse. Three human ApoE gene has 3 alleic variants on chromosome 19, i.e., ApoE2 (Cys112 and Cys158), ApoE3 (Cys112 and Arg158), and ApoE4 (Arg112 and Arg158), and ApoE4 generation is associated with increased risk of sporadic onset of AD [31]. ApoE is mainly produced and secreted by astrocytes in brain, and it stimulates Aβ uptake by neurons due to its affinity for Aβ [32]. Astrocytes carrying ApoE4 caused impaired degradation of Aβ in neurons and downregulation of synaptic proteins, which is cancelled by the genetic ablation of ApoE [32,33]. In this article, 2-AG or cadherin/catenin-mediated regulation of neurodegenerative process is mainly focused with the recent progress in pharmacological aspects, in addition to the topic on LRP-mediated clearance of Aβ.
Dysfunction of Cadherin/Catenin in AD
Synapse and spine morphology are maintained by several integral membrane proteins such as cadherins, or Rho family proteins [34]. Catenin family proteins interact with cadherins, but also regulate Rho family proteins, as seen in the inhibition of RhoA activity by p120 catenin [13]. p120 catenin binding to cadherin and RhoA inhibition are mutually exclusive, and overexpression of p120 catenin causes blanching morphology by inhibiting the guanine-nucleotide exchange activity of RhoA, which is then blunted by dominant active RhoA; this may indicate the involvement of p120 catenin in regulating cadherin function through indirect effects on RhoA-mediated cytoskeletal reorganization. p120 catenin regulates dendritic spine morphogenesis and innervation with the formation of synaptic clusters and axonal filopodia by modulating Rho GTPase activity, as seen during hippocampal neuronal development or formation of neuron-muscle junctions [14,35]. Additionally, δ-catenin interacts with p190 Rho guanine nucleotide exchange factor by selective competition with RhoA activity among Rho GTPases, promoting dendritogenesis and spine morphogenesis (Table 1) [36].
![]()
Table 1: Physiological roles of catenin family proteins in neuronal functions.
View Table 1
β-catenin interacts with the C-terminal domain of N-cadherin, mediating modulation of glutamatergic synaptic currents with homophilic N-cadherin adhesive activity. Although abnormal morphology of dendritic spines is observed by loss of β-catenin, this does not involve the actin cytoskeleton [37]. N- and E-cadherins reside at synaptic junctions in mutually exclusive patterns [3]. Either cadherin is necessary for PS1 interactions with β-catenin, thereby promoting degradation of β-catenin through the ubiquitin-proteasome system [6] (Figure 1). PS1/2 deficient cells exhibit accumulation of phospho-β-catenin with higher generation levels of reactive oxygen species (ROS) inducing cytotoxicity [9]. Amyloid precursor protein (APP), a type I membrane protein, also functions as a synaptic modulator and is proteolytically processed by β-secretase (BACE) and γ-secretase complexes, resulting in the generation of Aβ peptide [38]. Neuronal activity-dependent Aβ generation causes decreased synaptic transmission, whilst Swedish mutation of APP (APPSwe) severely depresses the transmission, presumably causing cognitive decline [4]. Interestingly, p120 catenins recruit γ-secretase to cadherins that promote their processing. These interactions inhibit the production of Aβ and intracellular domain of APP called AICD, suggesting that cadherin functions as one of the integral determinants of APP processing in neuronal homeostasis [7,8] (Figure 1). Interference of N-cadherin function by homophilic binding with N-terminal peptides, or expression of its ectodomain-shed C-terminal fragments, has been found to accelerate the effects of oligomeric Aβ on synaptic dysfunction [39]. Another catenin family protein, δ-catenin or neural plakophilin-related armadillo protein (NPRAP), interacts with PS1, inducing PS1 expression to suppress dendritic branching [40].
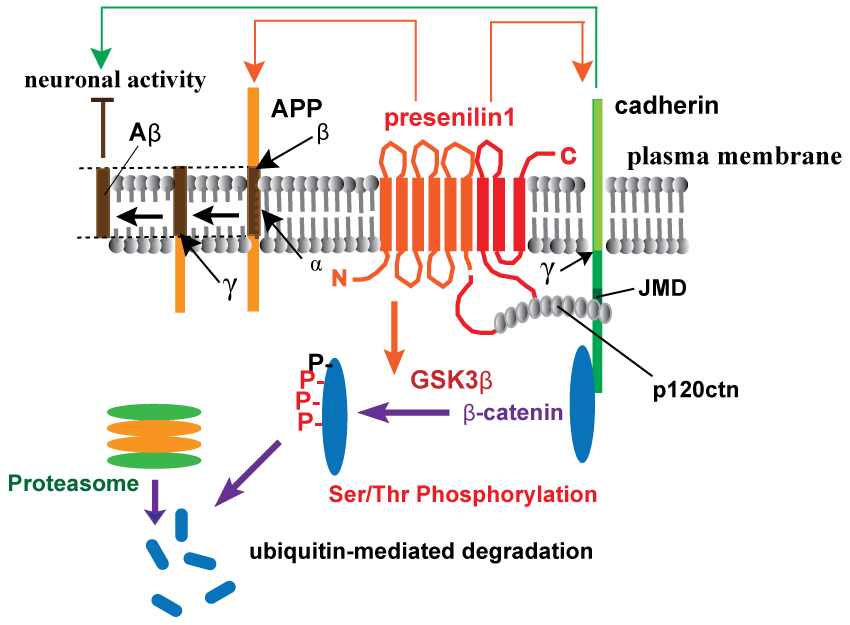
.
Figure 1: Schematic depiction of presenilin-1-mediated regulation of synaptic functions by cadherin/catenin complex.
Synaptic function is maintained by β-catenin in a cadherin-dependent manner, regulating synaptic strength and dendritic spine morphology [37]. Presenilin-1 (PS1) interacts with β-catenin through cadherins, regulating β-catenin stability by GSK3β-mediated Ser/Thr phosphorylation and ubiquitin-proteasomal degradation [6]. p120 catenin is required for cadherin-adhesive functions and also involved in the restrictive processing of cadherins through PS1 interaction via the JMD sequence. p120 catenin competes with amyloid precursor protein (APP) in its proteolysis catalyzed by the γ-secretase complex, including nicastrin, Pen2, and Aph1 (γ with an arrow in the figure indicates the cleavage site in APP or cadherin) [7,8]. Neural activity also controls APP processing and vice versa: imbalance in uncleaved APP levels and Aβ generation by familial Alzheimer's disease mutations can induce depression of synaptic transmission [4].
View Figure 1
Dysregulation of β-catenin in PD
Parkinson disease (PD) is a neurological disorder associated with selective degeneration of midbrain dopaminergic neurons and with gliosis. Activation of the Wnt/β-catenin pathway is required for the generation of dopaminergic neurons in the ventral midbrain but is also necessary for the prevention of cell death induced by 6-hydroxydopamine and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) with oxidative load [41]. Systemic administration of these agents induces astroglial activation, resulting in expression of Wnt1 and inflammatory cytokines, and induction of β-catenin signaling cascades to protect dopaminergic neurons from neurotoxicity [42]. Wnt1 secreted from ventral midbrain astrocytes injured by MPTP treatment induces β-catenin stabilization in mesencephalic neurons expressing dopamine transporters and wnt1 depletion nullifies the protection of tyrosine hydroxylase-positive (TH+) neurons. This survival pathway is regulated by Frizzled (Fzd)-mediated suppression of GSK-3β, which destabilizes β-catenin and activates caspase. β-Catenin then translocates into the nucleus to activate Wnt-responsive genes, or functions as a protective molecule against oxidative stress in the neurons [43]. Interestingly, MPTP treatment causes temporal suppression of neurogenesis in the subventricular zone (SVZ), as neurotoxic effects are correlated with microglial activation with increased NADPH-oxidase generating reactive oxygen species [11]. Impaired neurogenesis is associated with GSK-3β activation and β-catenin downregulation in neural progenitor cells. This process is modulated by the anti-inflammatory drug, HCT1026, which mitigates the microglial toxicity by blunting phagocyte oxidase activity, and activating PI3K/Akt and Nrf2 signaling [11,12]. Interestingly, treatment with Rho kinase inhibitors, such as fasudil, recovers TH+ neurons damaged by MPTP, accompanied with decreased inflammation and accumulation of Frizzled and β-catenin [44,45].
Imbalance of Lipid Signaling in AD
In healthy conditions, microglia maintain neuronal homeostasis through several transmembrane proteins such as CD200 and CD47 expressed in neurons as well as microglia, and their corresponding receptors CD200R and CD172 are expressed in microglia [46,47]. However, pathological neuronal damage induces the collapse of surveillance system engaged by these neuron/microglial interactions, and resulting microglia activation propagates alarm signals through the response of DAP12 adaptor proteins to a neuroinflammatory condition [46,48]. The expression of CD200 is regulated by inflammatory conditions: treatment of rats with Aβ (Aβ) decreases CD200 expression, whilst IL-4 directly upregulates the expression and attenuates Aβ-induced microglial activation [49]. Interestingly, DAP12 signaling in immune cells is indirectly linked to the regulation of Toll-like receptor (TLR) signaling, and crosstalk between TLRs and Aβ pathways have been elucidated in AD models [50,51]. CD14 that function as LPS receptor is known to interact with Aβ fibrils in physical proximity to TLR4 and mediate microglial activation and neurotoxicity [52]. Besides the microglial surveillance system, regulation of synaptic transmission by several lipids plays an additional critical role in maintaining neuronal homeostasis. PIP2 metabolism is important for the membrane trafficking and regulation of ion channels. Interestingly, FAD mutations of PS1, such as ΔE9 or L286V, and PS2 mutations N141I lowered PIP2 levels, inversely correlating well with Aβ42 levels and aberrant Mg2+-inhibited cation (MIC) channel activity [53]. γ-Secretase inhibitors did not affect generation of PIP2 turnover, suggesting that PIP2 itself is critical for the activity of γ-secretase and MIC/TPRM7 channels. Oligomeric Aβ42 also disrupted PIP2 levels and this effect was cancelled by the haploinsufficiency for synaptojanin 1, the primary brain PIP2 phosphatase in the synapses, suggesting that the PIP2 balance is important for suppression of Aβ-induced synaptic dysfunction [54].
ApoE-Mediated Aβ Clearance and Synaptic Regulation
ApoE4, which accounts for 15-20% of the population, is the primary genetic risk factor for sporadic and late-onset familial forms of AD although the ε2 allele of apoE is known to be associated with lower risk for AD [31]. LRP family proteins (LRP1, 2 or 5/6) promote clearance of Aβ, and ApoE is also involved in Aβ metabolism as shown by the efficacy of isoform-dependent complex formation between ApoE and Aβ. The interaction is related to inverse correlation with the risk of AD [55-57]. Aβ contains the binding site for ApoE and residues 12-28 of Aβ (Aβ12-28P) has been used for the interference of the association between ApoE and Aβ [32]. Astrocytes secreted ApoE and decreased the neuronal degradation of Aβ, resulting in increased production of neuronal Aβ oligomers and disruption of synaptic homeostasis [32]. Treatment with Aβ12-28P promoted the clearance of intracellular Aβ accumulation and nullified the loss of synaptic proteins such as NMDA receptor and synaptophysin. Comparative study using AD transgenic model harboring each ε allele of ApoE showed higher amounts of fibrillar Aβ in human ApoE4-expressing mouse than ApoE2 or ApoE3 transgenic mouse, suggesting that the interaction between ApoE4 and Aβ has detrimental effect on neuron/glia communication [32]. Mutant APP transgenic mice expressing ApoE4 increased synaptic activity in specific regions in their brains, which caused overproduction of Aβ and ubiquitin-positive dilated axons [58,59]. Human ApoE4 genotype itself has been shown to affect neurotransmission through changes in presynaptic terminal composition as seen in increased expression of vesicular glutamate transporter VGLUT1 in animal model [60]. Interestingly, ApoE4 has an inhibitory effect on Wnt signaling through LRP5 or very low density lipoproteins (VLDLs) that functions as Wnt co-receptors in cell-based analysis [61]. Alleic variants of LRP6 have also been implicated in putative genetic risk haplotype for late-onset AD associated with ApoE4 carriers from genome-wide linkage studies [62]. Substitution of Ile1062 to Val in LRP6, one of the genetic variations, downregulated β-catenin signaling in response to Wnt3a, suggesting the decreased Wnt/β-catenin signaling may be attributed to the neurodegenerative process.
Pathological Role of MAGL and PLA in AD
A lipidomic analysis of fatty acids of transgenic (TG) mice expressing human APP reveals increases in AA and its metabolites, such as prostaglandin E2 (PGE2), contributing to excitotoxicity [63]. These metabolic changes are attributed to the upregulation of group IV isoforms of phospholipase A2 (GIVA-PLA2) in the hippocampus of APP TG mice and patients with AD. Treatments with inhibitors of GIVA-PLA2 or arachidonyl trifluoromethyl ketone (AACOCF3) suppressed Aβ-induced neurotoxicity and genetic ablation of GIVA-PLA2 in APP TG mice, improving learning and memory defected [63]. Aβ and AA acutely increase the expression levels of AMPA receptor in neurons, whilst the AMPA-mediated Ca2+ influx may cause further activation of GIVA-PLA2, resulting in the Aβ-induced behavioral deficits in the APP TG mice [64]. Although GIVA-PLA2 targeting reagents have been considered promising for clinical intervention, the development of the specific chemicals has been a challenging issue [64].
Involvement of MAGL in dysregulated endocannabinoid metabolism has also been shown in several APP TG models. MAGL inactivation by genetic manipulation and treatment with MAGL-specific inhibitor JGL-184 has been found to suppress gliosis and cytokine production, such as IL-6 and TNFα, ameliorating the pathological process of Aβ deposition [26,27]. These anti-neuroinflammatory effects by MAGL inhibition are independent of CBR activation by 2-AG, as the treatments with antagonists of CBRs are unable to reverse the effects. Interestingly, transgenic mice carrying FAD mutations in APP and PS1 exhibited reductions in expressed levels of glutaminergic α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) or N-methyl-D-aspartate (NMDA) receptors in the cortex and hippocampus; whereas treatment with JZL-184 reversed the pathological effects, with maintaining the integrity of the synaptic morphology as shown in the improvement of spatial learning and memory [27]. In AD mouse models long-term JZL-184 treatment decreased BACE1 expression and Aβ/CTFβ generation in both the cortex and hippocampus [27]. The following molecular mechanism is further elucidated by aberrant MAGL activation downregulating PPARγ transcriptional activity through 2-AG degradation; MAGL activates NF-κB, resulting in BACE1 upregulation and Aβ-mediated neuroinflammation [65]. The beneficial effects of MAGL inhibition are independent of CBRs, suggesting that 2-AG may function as a direct activator for PPARγ-mediated transcription by crossing the plasma membrane, which implicates a novel mode of 2-AG action in maintenance of neuronal homeostasis.
2-AG Degradation System in Neuroinflammation
As for LPS-induced inflammation, selective LPS-mediated dopaminergic neurotoxicity is well known; low doses of LPS demonstrate toxicity in DA neurons in the presence of microglia in vitro, and result in delayed and progressive loss of DA neurons with precedent microglial activation in vivo [66]. This effect is in contrast to the acute neurotoxicity induced by MPTP or 6-OHDA; however, oxidative stress generated by microglia is also critical for the dopaminergic neurodegeneration [67]. Recently, the Cravatt group found that genetic MAGL inactivation, or treatment with JZL-184, accumulated AA, but not AEA, and led to no microglial activation when treated with LPS. These findings suggest that microglial MAGL might play a central role in the regulation of 2-AG homeostasis in inflammatory conditions [25]. However, microglial MAGL inactivation by lentiviral RNAi or JZL184 treatment in culture conditions, as well as MAGL introduction into immortalized microglial BV-2 cell lines lacking MAGL expression, did not affect the production of inflammatory cytokines by LPS treatment (Figure 2) [29]. Microglial MAGL was transcriptionally downregulated but stabilized by LPS treatment promoting 2-AG degradation, suggesting that microglial MAGL is not explicitly required for the generation of inflammatory cytokines. Recently, Viader et al. reported that astroglial MAGL contributed predominantly to neuroinflammatory responses by inducing 2-AG hydrolysis, resulting in AA production. Additionally, astrocyte-specific inactivation of MAGL significantly affected cytokine production in the presence of LPS, suggesting that 2-AG homeostasis may be primarily regulated by neurons and astrocytes in inflammatory conditions, whilst generated AA or prostaglandins (PG) regulate microglial activation during the neurodegenerative process [68]. In MPTP-induced dopaminergic neurodegeneration models, JZL184 treatment or MAGL knockout caused significant suppression of the production of AA or PG [25], suggesting that similar molecular mechanisms might be commonly involved in the neurodegeneration process in PD model.
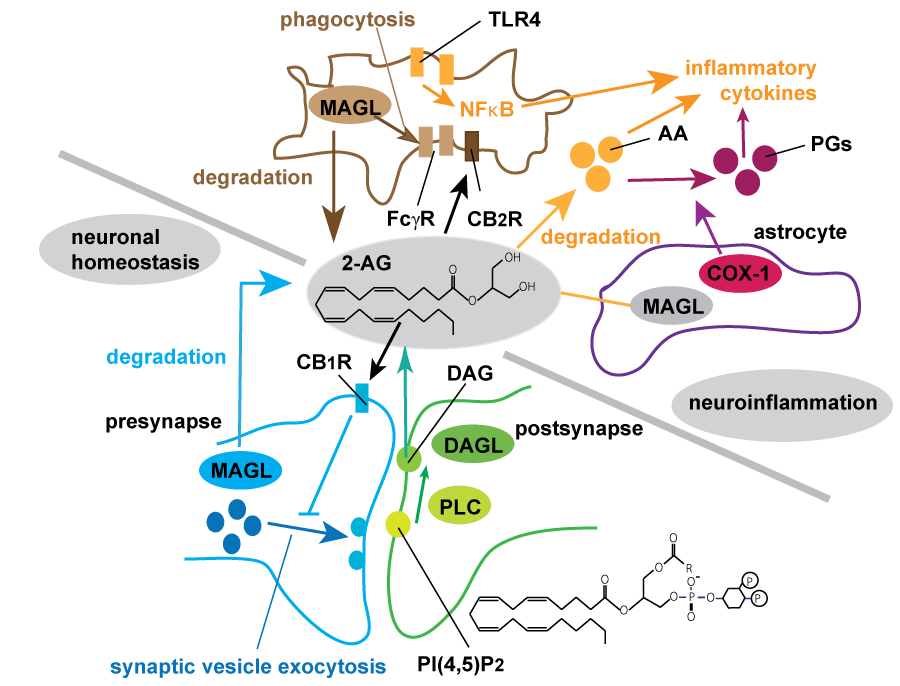
.
Figure 2: Summary of endocannabinoid biosynthesis and degradation regulating synaptic transmission and neuroinflammation.
2-arachidonoylglycerol (2-AG), the most abundant endocannabinoid, is generated in postsynaptic compartment by hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) by PLC followed by degradation of DAG by sn-1-specific diacylglycerol lipases (DAGLβ and DAGLβ). Generated 2-AG retrogradely acts as a ligand for CB1 receptor (CB1R) which resides in presynaptic compartment, and suppresses the synaptic transmission known as depolarization-induced suppression of excitation (DSE) and inhibition (DSI) [20,21]. MAGL hydrolyzes most of 2-AG in the brain, resulting in the termination of 2-AG mediated CBR signaling [23]. In neuroinflammation neuron and astrocyte-derived MAGL mostly maintain 2-AG homeostasis and astrocytic MAGL is mainly contributed to the production of prostaglandins (PGs) and inflammatory cytokines such as IL-6 or TNF [68]. Multiple pathways utilizing arachidnoic acid (AA) may be involved in microglial activation during neurodegenerative process. Microglial MAGL promotes Fcγ-mediated phagocytosis and uptake of Aβ in neuroinflammatory conditions [28,29].
View Figure 2
Conclusion
Catenin proteins have been found to mediate diverse cellular signaling patterns, including synaptic responses and dendritic spine morphology both necessary for neuronal homeostasis. Enhanced Aβ production is seen to impair synaptic function, also pathologically is linked to dysfunctional cadherin-mediated adhesion and detrimental effects of ApoE4. Furthermore, PS1 is integrated with cadherin-mediated catenin signaling and regulation of cadherin and APP processing, by a mutually restrictive mechanism. Aberrant γ-secretase activity caused by FAD PS mutations can result in decreased neuronal activity and synaptic depression, in addition to affect APP processing via PS mutations [4,38,40]. AMPA and NMDA-mediated transmission are downregulated by APP mutations accompanied by MAGL activation, with the latter's involvement previously identified by JZL184-dependent restoration of stability in animal AD models [4,27]. MAGL has a predominant role in downregulation of 2-AG signaling and catalyzes the production of AA and PGs in neuroinflammatory conditions; however, MAGL activity affects several signaling processes regulated by NF-κB or PPARγ, dependent on the context of neurodegeneration [65]. In neuroinflammation models with MPTP or LPS administration, MAGL is critical for microglial activation, but neither cell-autonomous MAGL activity, nor NF-κB activation in microglia is required [25,29]. Recent progress on these studies has highlighted the importance of neuronal and astrocytic MAGL in AD and PD models, respectively. Based on these findings, further elucidation of unknown critical signaling pathways affected by 2-AG turnover in each neurodegeneration model would be crucial for future development of clinical interventions.
Acknowledgement
I thank Dr. N. K. Robakis at Mount Sinai School of Medicine in New York for permission to mention results of our collaborative work.
References
-
Nägerl UV, Eberhorn N, Cambridge SB, Bonhoeffer T (2004) Bidirectional activity-dependent morphological plasticity in hippocampal neurons. Neuron 44: 759-767.
-
Yu X, Malenka RC (2003) Beta-catenin is critical for dendritic morphogenesis. Nat Neurosci 6: 1169-1177.
-
Fannon AM, Colman DR (1996) A model for central synaptic junctional complex formation based on the differential adhesive specificities of the cadherins. Neuron 17: 423-434.
-
Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, et al. (2003) APP processing and synaptic function. Neuron 37: 925-937.
-
Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, et al. (2003) Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39: 409-421.
-
Serban G, Kouchi Z, Baki L, Georgakopoulos A, Litterst CM, et al. (2005) Cadherins mediate both the association between PS1 and beta-catenin and the effects of PS1 on beta-catenin stability. J Biol Chem 280: 36007-36012.
-
Kouchi Z, Barthet G, Serban G, Georgakopoulos A, Shioi J, et al. (2009) p120 catenin recruits cadherins to gamma-secretase and inhibits production of Abeta peptide. J Biol Chem 284: 1954-1961.
-
Barthet G, Georgakopoulos A, Robakis NK (2012) Cellular mechanisms of gamma-secretase substrate selection, processing and toxicity. Prog Neurobiol 98: 166-175.
-
Boo JH, Song H, Kim JE, Kang DE, Mook-Jung I (2009) Accumulation of phosphorylated beta-catenin enhances ROS-induced cell death in presenilin-deficient cells. PLoS One 4: e4172.
-
Matter C, Pribadi M, Liu X, Trachtenberg JT (2009) Delta-catenin is required for the maintenance of neural structure and function in mature cortex in vivo. Neuron 64: 320-327.
-
L'Episcopo F, Tirolo C, Testa N, Caniglia S, Morale MC, et al. (2012) Plasticity of subventricular zone neuroprogenitors in MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) mouse model of Parkinson's disease involves cross talk between inflammatory and Wnt/beta-catenin signaling pathways: functional consequences for neuroprotection and repair. J. Neurosci 32: 2062-2085.
-
L'Episcopo F, Tirolo C, Testa N, Caniglia S, Morale MC, et al. (2013) Aging-induced Nrf2-ARE pathway disruption in the subventricular zone drives neurogenic impairment in parkinsonian mice via PI3K-Wnt/beta-catenin dysregulation. J Neurosci 33: 1462-1485.
-
Anastasiadis PZ, Moon SY, Thoreson MA, Mariner DJ, Crawford HC, et al. (2000) Inhibition of RhoA by p120 catenin. Nat Cell Biol 2: 637-644.
-
Elia LP, Yamamoto M, Zang K, Reichardt LF (2006) p120 catenin regulates dendritic spine and synapse development through Rho-family GTPase and cadherins. Neuron 51: 43-56.
-
Israely I, Costa RM, Xie CW, Silva AJ, Kosik KS, et al. (2004) Deletion of the neuron-specific protein delta-catenin leads to severe cognitive and synaptic dysfunction. Curr. Biol. 14: 1657-1663.
-
Bamji SX, Shimazu K, Kimes N, Huelsken J, Birchmeier W, et al. (2003) Role of beta-catenin in synaptic vesicle localization and presynaptic assembly. Neuron 40: 719-731.
-
Medina M, Marinescu RC, Overhauser J, Kosik KS (2000) Hemizygosity of delta-catenin (CTNND2) is associated with severe mental retardation in Cri-du-Chat syndrome. Genomics 63: 157-164.
-
Micheva KD, Holz RW, Smith SJ (2001) Regulation of presynaptic phosphatidylinositol 4,5-biphosphate by neuronal activity. J Cell Biol 154: 355-368.
-
Di Paolo G, Moskowitz HS, Gipson K, Wenk MR, Voronov S, et al. (2004) Impaired PtdIns(4,5)P2 synthesis in nerve terminals produces defects in synaptic vesicle trafficking. Nature 431: 415-422.
-
Tanimura A, Yamazaki M, Hashimotodani Y, Uchigashima M, Kawata S, et al. (2010) The endocannabinoid 2-arachidonoylglycerol produced by diacylglycerol lipase alpha mediates retrograde suppression of synaptic transmission. Neuron 65: 320-327.
-
Hashimotodani Y, Ohno-Shosaku T, Tsubokawa H, Ogata H, Emoto, K et al. (2005) Phospholipase Cbeta serves as a coincidence detector through its Ca2+ dependency for triggering retrograde endocannabinoid signal. Neuron 45: 257-268.
-
Savinainen JR, Saario SM, Laitinen JT (2012) The serine hydrolases MAGL, ABHD6 and ABHD12 as guardians of 2-arachidonoylglycerol signalling through cannabinoid receptors. Acta Physiol (Oxf) 204: 267-276.
-
Hashimotodani Y, Ohno-Shosaku T, Kano M (2007) Presynaptic monoacylglycerol lipase activity determines basal endocannabinoid tone and terminates retrograde endocannabinoid signaling in the hippocampus. J. Neurosci. 31: 1211-1219.
-
Witting A, Walter L, Wacker J, Möller T, Stella N (2004) P2X7 receptors control 2-arachidonoylglycerol production by microglial cells. Proc Natl Acad Sci U S A 101: 3214-3219.
-
Nomura DK, Morrison BE, Blankman JL, Long JZ, Kinsey SG, et al. (2011) Endocannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science 334: 809-813.
-
Piro JR, Benjamin DI, Duerr JM, Pi Y, Gonzales C (2012) A dysregulated endocannabinoid-eicosanoid network supports pathogenesis in a mouse model of Alzheimer's disease. Cell Rep. 1: 617-623.
-
Chen R, Zhang J, Wu Y, Wang D, Feng G, et al. (2012) Monoacylglycerol lipase is a therapeutic target for Alzheimer's disease. Cell Rep 2: 1329-1339.
-
Mulder J, Zilberter M, Pasquaré SJ, Alpár A, Schulte G, et al. (2011) Molecular reorganization of endocannabinoid signalling in Alzheimer's disease. Brain 134: 1041-1060.
-
Kouchi Z (2015) Monoacylglycerol lipase promote Fcgamma receptor-mediated phagocytosis in microglia but does not regulate LPS-induced upregulation of inflammatory cytokines. Biochem. Biophys. Res. Commun. 464: 603-610.
-
Walter L, Franklin A, Witting A, Wade C, Xie Y, et al. (2003) Nonpsychotropic cannabinoid receptors regulate microglial cell migration. J Neurosci 23: 1398-1405.
-
Schmechel DE, Saunders AM, Strittmatter WJ, Crain BJ, Hulette CM, et al. (1993) Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci U S A 90: 9649-9653.
-
Kuszczyk MA, Sanchez S, Pankiewicz J, Kim J, Duszczyk M, et al. (2013) Blocking the interaction between Apolipoprotein E and Abeta reduces intraneuronal accumulation of Abeta and inhibits synaptic degeneration. Am J. Pathol. 182: 1750-1768.
-
Sadowski M, Pankiewicz J, Scholtzova H, Ripellino JA, Li Y, et al. (2004) A synthetic peptide blocking the Apolipoprotein E/beta-amyloid binding mitigates beta-amyloid toxicity and fibril formation in vitro and reduces beta-amyloid plaques in transgenic mice. Am J. Pathol. 165: 937-948.
-
Arikkath J, Reichardt LF (2008) Cadherins and catenins at synapses: roles in synaptogenesis and synaptic plasticity. Trends Neurosci 31: 487-494.
-
Chen C, Li PP, Madhavan R, Peng HB (2012) The function of p120 catenin in filopodial growth and synaptic vesicle clustering in neurons. Mol Biol Cell 23: 2680-2691.
-
Kim H, Han JR, Park J, Oh M, James SE, et al. (2008) Delta-Catenin-induced dendritic morphogenesis. An essential role of p190RhoGEF interaction through Akt1-mediated phosphorylation. J Biol Chem 283: 977-987.
-
Okuda T, Yu LM, Cingolani LA, Kemler R, Goda Y (2007) Beta-Catenin regulates excitatory postsynaptic strength at hippocampal synapses. Proc Natl Acad Sci U S A 104: 13479-13484.
-
Robakis NK (2014) Cell signaling abnormalities may drive neurodegeneration in familial Alzheimer disease. Neurochem Res 39: 570-575.
-
Andreyeva A, Nieweg K, Horstmann K, Klapper S, Müller-Schiffmann A, et al. (2012) C-terminal fragment of N-cadherin accelerates synapse destabilization by amyloid-beta. Brain 135: 2140-2154.
-
Kim JS, Bareiss S, Kim KK, Tatum R, Han JR, et al. (2006) Presenilin-1 inhibits delta-catenin-induced cellular branching and promotes delta-catenin processing and turnover. Biochem Biophys Res Commun 351: 903-908.
-
L'Episcopo F, Tirolo C, Testa N, Caniglia S, Morale MC, et al. (2011) Reactive astrocytes and Wnt/beta-catenin signaling link nigrostriatal injury to repair in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson's disease. Neurobiol Dis 41: 508-527.
-
L'Episcopo F, Serapide MF, Tirolo C, Testa N, Caniglia S, et al. (2011) A wnt1 regulated Frizzled-1/beta-catenin signaling pathway as a candidate regulatory circuit controlling mesencephalic dominergic neuron-astrocyte crosstalk: Therapeutical relevance for neuron survival and neuroprotection. Mol. Neurodegeneration 6: 49.
-
Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, et al. (2008) A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci 28: 264-278.
-
Tönges L, Frank T, Tatenhorst L, Saal KA, Koch JC, et al. (2012) Inhibition of rho kinase enhances survival of dopaminergic neurons and attenuates axonal loss in a mouse model of Parkinson's disease. Brain 135: 3355-3370.
-
Zhao Y, Zhang Q, Xi J, Xiao B, Li Y, et al. (2015) Neuroprotective effect of fasudil on inflammation through PI3K/Akt and Wnt/b-catenin dependent pathways in a mice model of Parkinson's disease. Int J Clin Exp Pathol 8: 2354-2364.
-
Hoek RM, Ruuls SR, Murphy CA, Wright GJ, Goddard R, et al. (2000) Down-regulation of the macrophage lineage through interaction with OX2 (CD200). Science 290: 1768-1771.
-
Koning N, Bö L, Hoek RM, Huitinga I (2007) Downregulation of macrophage inhibitory molecules in multiple sclerosis lesions. Ann Neurol 62: 504-514.
-
Takahashi K, Rochford CD, Neumann H (2005) Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med 201: 647-657.
-
Lyons A, Downer EJ, Crotty S, Nolan YM, Mills KH, et al. (2007) CD200 ligand receptor interaction modulates microglial activation in vivo and in vitro: a role for IL-4. J Neurosci 27: 8309-8313.
-
Hamerman JA, Tchao NK, Lowell CA, Lanier LL (2005) Enhanced Toll-like receptor responses in the absence of signaling adaptor DAP12. Nat Immunol 6: 579-586.
-
Bakker AB, Hoek RM, Cerwenka A, Blom B, Lucian L, et al. (2000) DAP12-deficient mice fail to develop autoimmunity due to impaired antigen priming. Immunity 13: 345-353.
-
Fassbender K, Walter S, Kühl S, Landmann R, Ishii K, et al. (2004) The LPS receptor (CD14) links innate immunity with Alzheimer's disease. FASEB J 18: 203-205.
-
Landman N, Jeong SY, Shin SY, Voronov SV, Serban G, et al. (2006) Presenilin mutations linked to familial Alzheimer's disease cause an imbalance in phosphatidylinositol 4,5-bisphosphate metabolism. Proc. Natl. Acad. Sci. 103: 19524-19529.
-
Berman DE, Dall'Armi C, Voronov SV, McIntire LB, Zhang H, et al. (2008) Oligomeric amyloid-beta peptide disrupts phosphatidylinositol-4,5-bisphosphate metabolism. Nat Neurosci 11: 547-554.
-
Tokuda T, Calero M, Matsubara E, Vidal R, Kumar A, et al. (2000) Lipidation of apolipoprotein E influences its isoform-specific interaction with Alzheimer's amyloid beta peptides. Biochem J 348 Pt 2: 359-365.
-
Kanekiyo T, Cirrito JR, Liu CC, Shinohara M, Li J, et al. (2013) Neuronal clearance of amyloid-beta by endocytic receptor LRP1. J Neurosci 33: 19276-19283.
-
Zerbinatti CV, Wahrle SE, Kim H, Cam JA, Bales K, et al. (2006) Apolipoprotein E and low density lipoprotein receptor-related protein facilitate intraneuronal Abeta42 accumulation in amyloid model mice. J Biol Chem 281: 36180-36186.
-
Tesseur I, Van Dorpe J, Bruynseels K, Bronfman F, Sciot R, et al. (2000) Prominent axonopathy and disruption of axonal transport in transgenic mice expressing human apolipoprotein E4 in neurons of brain and spinal cord. Am J Pathol 157: 1495-1510.
-
Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, et al. (2011) Human apoE isoforms differentially regulate brain amyloi beta peptide clearance. Sci Transl Med 3: 89ra57.
-
Dumanis SB, DiBattista AM, Miessau M, Moussa CE, Rebeck GW (2013) APOE genotype affects the pre-synaptic compartment of glutamatergic nerve terminals. J Neurochem 124: 4-14.
-
Caruso A, Motolese M, Iacovelli L, Caraci F, Copani A, et al. (2006) Inhibition of the canonical Wnt signaling pathway by apolipoprotein E4 in PC12 cells. J Neurochem 98: 364-371.
-
De Ferrari GV, Papassotiropoulos A, Biechele T, De-Vrieze FW, Avila ME, et al. (2007) Common genetic variation within the low-density lipoprotein receptor-related protein 6 and late-onset Alzheimer's disease. Proc. Natl. Acad. Sci. 104: 9439-9439.
-
Sanchez-Mejia RO, Newman JW, Toh S, Yu GQ, Zhou Y, et al. (2008) Phospholipase A2 reduction ameliorates cognitive deficits in a mouse model of Alzheimer's disease. Nat Neurosci 11: 1311-1318.
-
Sanchez-Mejia RO, Mucke L (2010) Phospholipase A2 and arachidonic acid in Alzheimer's disease. Biochim Biophys Acta 1801: 784-790.
-
Zhang J, Hu M, Teng Z, Tang YP, Chen C3 (2014) Synaptic and cognitive improvements by inhibition of 2-AG metabolism are through upregulation of microRNA-188-3p in a mouse model of Alzheimer's disease. J Neurosci 34: 14919-14933.
-
Block ML, Hong JS (2005) Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol 76: 77-98.
-
Gao HM, Liu B, Zhang W, Hong JS (2003) Critical role of microglial NADPH oxidase-derived free radicals in the in vitro MPTP model of Parkinson's disease. FASEB J 17: 1954-1956.
-
Viader A, Blankman JL, Zhong P, Liu X, Schlosburg JE, et al. (2015) Metabolic interplay between astrocytes and neurons regulates endocannabinoid action. Cell Rep 12: 798-808.
-
Abe K, Chisaka O, Van Roy F, Takeichi M (2004) Stability of dendritic spines and synaptic contacts is controlled by alpha N-catenin. Nat Neurosci 7: 357-363.
-
Nowotarski SH, Peifer M (2014) Cell biology: a tense but good day for actin at cell-cell junctions. Curr Biol 24: R688-690.
-
Sun Y, Aiga M, Yoshida E, Humbert PO, Bamji SX (2009) Scribble interacts with beta-catenin to localize synaptic vesicles to synapses. Mol Biol Cell 20: 3390-3400.
-
Schuman EM, Murase S (2003) Cadherins and synaptic plasticity: activity-dependent cyclin-dependent kinase 5 regulation of synaptic beta-catenin-cadherin interactions. Phil. Trans. R. Soc. Lond. B Biol Sci 358: 749-756.
-
Abu-Elneel K, Ochiishi T, Medina M, Remedi M, Gastaldi L, et al. (2008) A delta-catenin signaling pathway leading to dendritic protrusions. J Biol Chem 283: 32781-32791.
-
Kim H, Han JR, Park J, Oh M, James SE, et al. (2008) Delta-catenin-induced dendritic morphogenesis. An essential role of p190RhoGEF interaction through Akt1-mediated phosphorylation. J Biol Chem 283: 977-987.
-
Davis MA, Ireton RC, Reynolds AB (2003) A core function for p120-catenin in cadherin turnover. J Cell Biol 163: 525-534.
-
Li W, Li Y, Gao FB (2005) Abelson, enabled, and p120 catenin exert distinct effects on dendritic morphogenesis in Drosophila. Dev Dyn 234: 512-522.
-
Piedra J, Miravet S, Castaño J, Pálmer HG, Heisterkamp N, et al. (2003) p120 Catenin-associated Fer and Fyn tyrosine kinases regulate beta-catenin Tyr-142 phosphorylation and beta-catenin-alpha-catenin Interaction. Mol Cell Biol 23: 2287-2297.
-
Boguslavsky S, Grosheva I, Landau E, Shtutman M, Cohen M, et al. (2007) p120 catenin regulates lamellipodial dynamics and cell adhesion in cooperation with cortactin. Proc Natl Acad Sci U S A 104: 10882-10887.
-
Wildenberg GA, Dohn MR, Carnahan RH, Davis MA, Lobdell NA, et al. (2006) p120-catenin and p190RhoGAP regulate cell-cell adhesion by coordinating antagonism between Rac and Rho. Cell 127: 1027-1039.
-
Nolze A, Schneider J, Keil R, Lederer M, Hüttelmaier S, et al. (2013) FMRP regulates actin filament organization via the armadillo protein p0071. RNA 19: 1483-1496.
-
Stahl B, Diehlmann A, Südhof TC (1999) Direct interaction of Alzheimer's disease-related presenilin 1 with armadillo protein p0071. J Biol Chem 274: 9141-9148.
-
Hatzfeld M, Green KJ, Sauter H (2003) Targeting of p0071 to desmosomes and adherens junctions is mediated by different protein domains. J Cell Sci 116: 1219-1233.
