

Journal of Dermatology Research and Therapy
Targeted Prodrug Design for the Treatment of Malignant Melanoma
Marcella Gabrielle Mendes Machado, Paulo Renato Yamasaki, Jean Leandro dos Santos and Chung Man Chin*
Faculdade de Ciências Farmacêuticas, UNESP - Univ Estadual Paulista, Departamento de Fármacos e Medicamentos
*Corresponding author:
Chung Man Chin, Faculdade de Ciências Farmacêuticas, UNESP - Univ Estadual Paulista, Departamento de Fármacos e Medicamentos. Rodovia Araraquara-Jaú Km 1, Cep 14800-903, Araraquara, SP, Brazil, E-mail: chungmc@fcfar.unesp.br
J Dermatol Res Ther, JDRT-2-019, (Volume 2, Issue 2), Short Review; ISSN: 2469-5750
Received: March 07, 2016 | Accepted: April 01, 2016 | Published: April 04, 2016
Citation: Machado MGM, Yamasaki PR, dos Santos JL, Chin CM (2016) Targeted Prodrug Design for the Treatment of Malignant Melanoma. J Dermatol Res Ther 2:019. 10.23937/2469-5750/1510019
Copyright: © 2016 Machado MGM, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Malignant melanoma is a serious health problem once the current chemotherapy exhibits resistance to traditional drugs. Several challenges must be overcome during drug design in order to increase the efficacy and safety of the new drugs. Therefore, the specific chemical release of cytotoxic agents near to the target is an attractive approach to improve the anticancer activity and reduce the systemic toxicity. Herein, this review article describes the advances in the development of targeted prodrugs for the treatment of malignant melanoma.
Keywords
Prodrug, Skin cancer, Melanoma
Introduction
Advanced malignant melanoma is a highly aggressive form of skin cancer, characterized to be one of the toughest treatments among human tumors [1,2]. In 2016, it has been estimated 76,000 new cases, responsible to cause 10,000 deaths only in the United States [3].
An increased number in resistance cases by malignant cells against the current therapy justifies the discovery of new drugs [4,5]. Dacarbazine was one of the first drug approved by US Food and Drug Administration (FDA) for melanoma treatment. This drug is used alone or in combination with other cytostatic agents and exhibits response rates ranging from 30-40% [6,7].
Nowadays, the current chemotherapy includes drugs, such as: a) temozolomide - used alone or through two different combinations [cisplatin, vinblastine and dacarbazine (CVD) or cisplatin, dacarbazine, carmustine, and tamoxifen (Dartmouth regimen)]; b) carboplatin and paclitaxel (sometimes combined with sorafenib); and c) immunotherapeutic drugs (ipilimumab, trametinib, pembrolizumab, vemurafenib, dabrafenib, peginterferon alfa-2b) [8]. In addition, targeted therapies for advanced melanoma involving BRAF kinase inhibitors and anti-cytotoxic T-lymphocyte antigen-4 (CTLA-4) antibodies have gained particular interest of researchers [9].
The molecular modification has demonstrated to be a promising approach to discover new anticancer drugs. Specifically, the development of prodrugs is an interesting strategy to overcome adverse effects and increase the drug specificity against malignant cells [10].
The term prodrug was first defined in 1958 by Adrien Albert [11] as an inactive compound that need an enzymatic or chemical biotransformation in vivo to release the active drug. By analogy, this strategy can be compared to a "Trojan horse" in that, the drug is a non-toxic compound per se; however, after chemical or enzymatic bioconversion, the active drug is released near and/or at the site of action (Figure 1) [10].
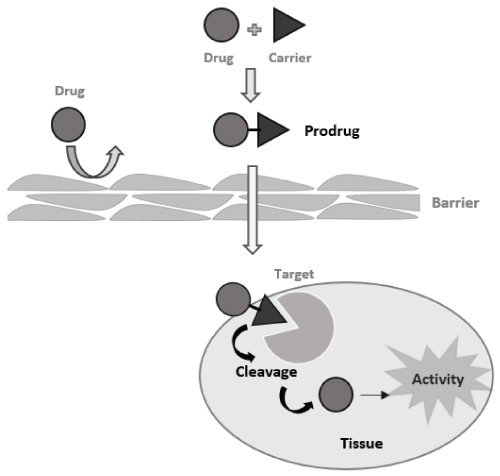
.
Figure 1: Prodrug strategy. The drug is released after chemical or enzymatic activation proving the pharmacological effect near to the site of action.
View Figure 1
There are two main categories of prodrugs: classical prodrugs and bioprecursors. For classical prodrugs, the active drug is linked to a carrier (small molecules or macromolecules) through a bioreversible covalent linkage [12]. Prodrug specificity can be obtained using carriers or linkers that can be identified by the target (membrane receptor or enzyme). This mechanism increases the drug efficacy and decrease in toxicity [12-19].
Capecitabine (1), an antineoplastic prodrug of 5-fluorouracil (5-FU) (2), is an example of classical prodrug (Figure 2). The active drug (5-FU) is released selectively into tumors cells by bioconversion of three enzymes (carboxylesterase, cytidine deaminase and thymidine phosphorylase) [20].
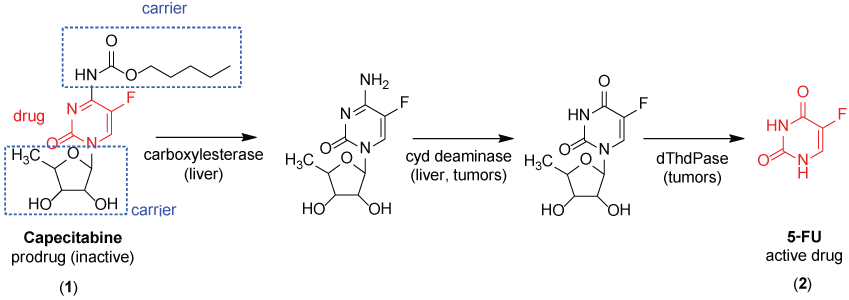
.
Figure 2: Structure and metabolic pathway of capecitabine (1). Cyd deaminase = cytidine deaminase; dThdPase = thymidine phosphorylase.
View Figure 2
On the other hand, bioprecursors prodrugs also need activation by metabolic processes in vivo; however they do not contain a carrier. Examples of bioprecursors include: statins (e.g., simvastatin), antiviral drugs (e.g., zidovudine and acyclovir) and chemotherapeutic drugs (e.g., tirapazamine) [12]. Figure 3 shows the chemical structure of cyclophosphamide (3), an antitumor bioprecursor that is metabolized in vivo by cytochrome P450 enzymes into active and inactive metabolites. For this example, the phosphoramide mustard (6) is the active compound with cytotoxic activity [21].
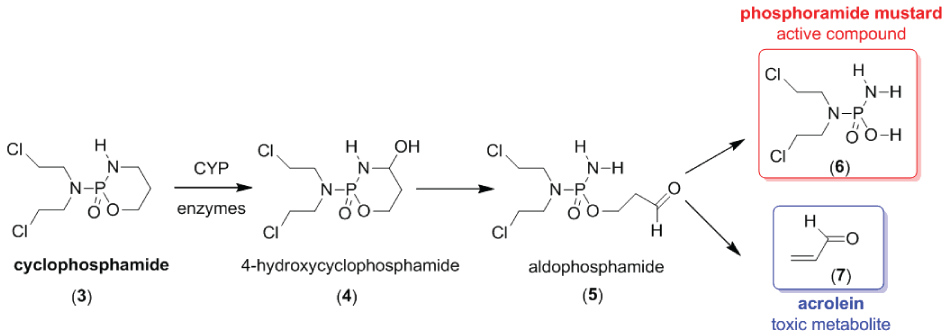
.
Figure 3: Activation of cyclophosphamide (3), an antitumor drug, to its active and inactive metabolites.
View Figure 3
Temozolomide (8) shown in Figure 4 as well as cyclophosphamide is an alkylating agent bioprecursor prodrug used in melanoma treatment (but not exclusively). The mechanism of action is to release a highly reactive methyldiazonium cation (11) that methylate DNA at N7 positions of guanine (preferentially), N3 adenine and O6 guanine residues [22-24].
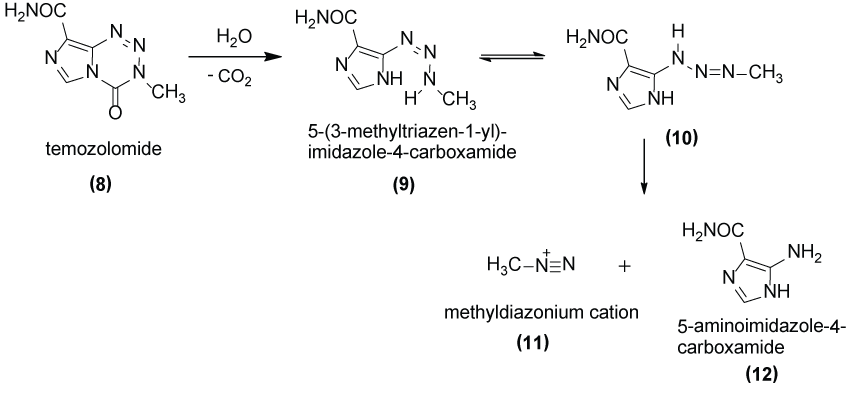
In this review article, our aim is to describe the use of the prodrug approach as a tool to discover new drugs for malignant melanoma. Therefore, it will be discussed the current advances and the strategies reported in the literature in the last years.
Prodrugs Useful to Treat Malignant Melanoma
Several endogenous enzymes have shown ability to activate prodrugs with antitumor activity. These enzymes can be exclusively expressed in tumors tissues or only overexpressed in malignant cells [12].
Tyrosinase, for example, is an enzyme found in melanocytes that shows an important function in melanogenesis. Mutant melanocytes demonstrate increased levels of tyrosinase compared to normal cells. Therefore, this enzyme can be explored as high selective in-situ tool for the activation of anti-melanoma Prodrugs [1,2].
Researchers have used tyrosinase substrates as chemotherapeutic drug carriers during the drug design of the prodrug MDEPT (Melanocyte-directed Enzyme Prodrug Therapy) [6,25]. The prodrug molecule was designed using a cytotoxic agent (drug) linked to a tyrosinase substrate (carrier). Inside the melanoma, after bioaction by tyrosinase, the active drug is released through an intramolecular reaction (Figure 5) [1]. Furthermore, additional mechanisms involving the formation of radical oxygen species (ROS) and glutathione reductase (GSH) depletion potentiates the drug toxicity inside the melanoma cells.
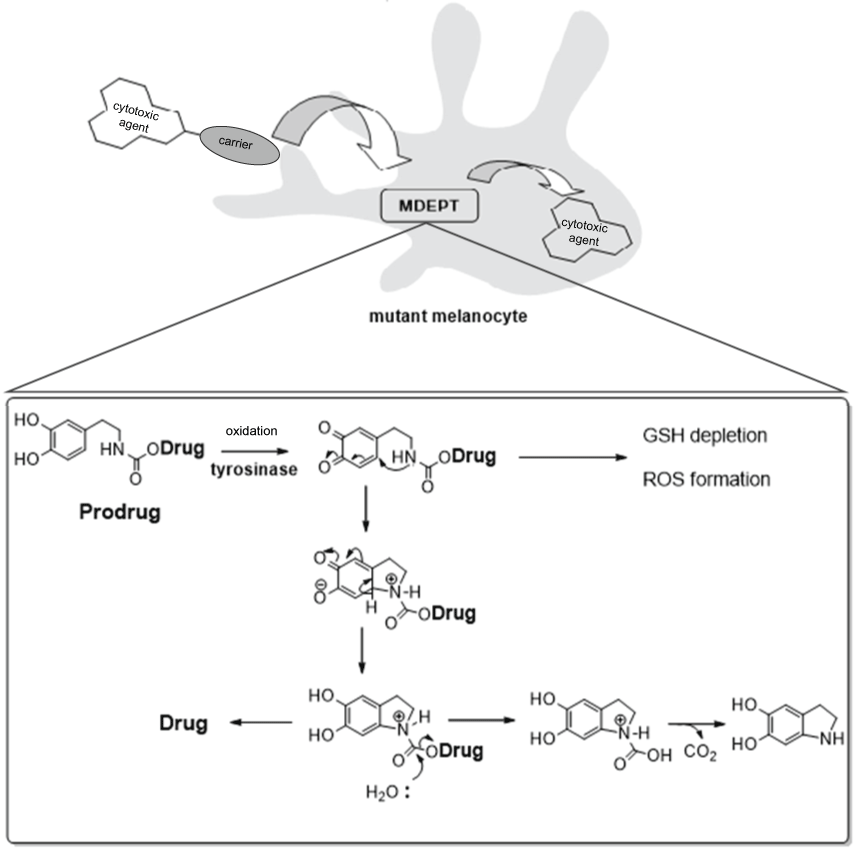
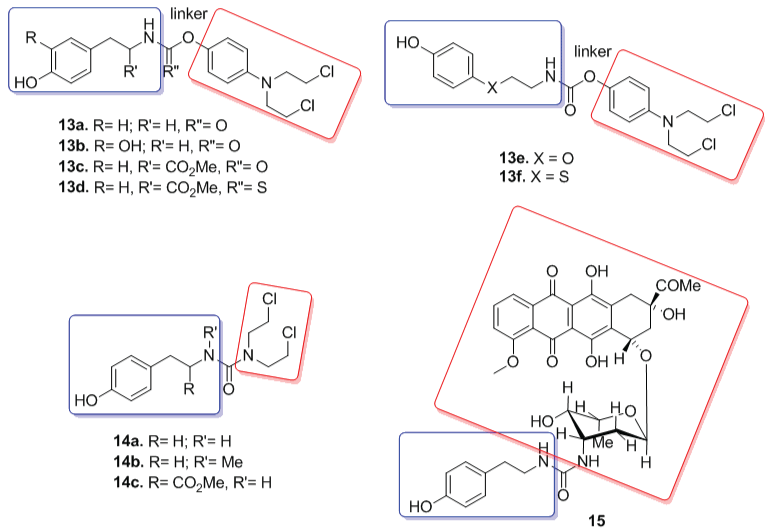
Jordan and co-workers [25] synthesized a series of prodrugs for MDEPT using as cytotoxic agents phenyl mustard (13a-13f), bisethyl amine mustard (14a-14c) and daunomycin (15). For all those compounds, it was used a carbamate, urea or thiourea linker (Figure 6). These compounds were activated by tyrosinase in the same way as described.
In order to evaluate the ability of all compounds to be activated by tyrosinase, the researchers used the enzyme purified of mushrooms. The reason for that is due to structural similarity since the active sites of both contain a common binuclear copper centre. In addition, human tyrosinase is not commercially available and several studies have reported mushroom's tyrosinase as a suitable and accurate model in earlier studies [26]. In this study, the experiments measured the rate of oxidation by tyrosinase (R). The maximum value of R (Rmax) correlates with the ability to release of the drug after oxidation.
Interestingly, it was observed a decreased rate in tyrosinase oxidation when the carbamate group (13c) is replaced by thiocarbamate (13d). The results also showed that compound 14a (R max = 20 nanomol/min) displayed superior oxidation rate compared to tyrosine methyl ester (R max = 17.5 nanomol/min), a natural substrate for tyrosinase. Moreover, compounds 13a, 13b and 14c (Figure 6) exhibited similar oxidation to tyrosine methyl ester; while 13e and 13f, containing a heteroatom insertion, were less effective in this experiment (Rmax = 7.5 nmol/min) [19].
.
Figure 6: Phenyl mustard prodrugs (13a-13f), bis-(2-chloroethyl)amine urea mustards (14a-14c) and daunomycin prodrug (15). In blue are represented chemical groups that act as tyrosinase substrates. After enzymatic activation, the active drugs (red) are released in order to kill the tumor cells.
View Figure 6
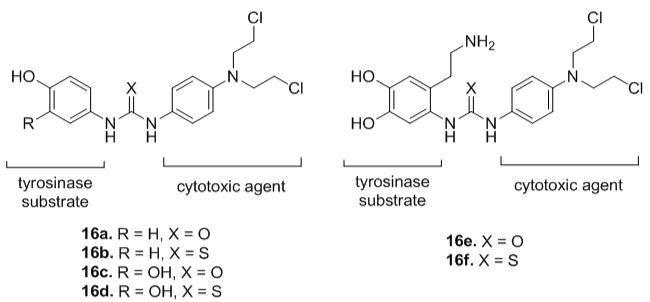
Knaggs et al. [27] obtained prodrugs (16a-16f) derived from 6-aminodopamine and 4-aminophenol as candidates for MDEPT (Figure 7). The compounds 16a-16d were oxidized at levels ranging from 70-78% of the rate compared to L-tyrosine (Rmax = 17 nmols/min), a natural substrate for tyrosinase. Compounds 16e and 16f have shown low rate of oxidation at levels of 50% and 25%, respectively. Drug release from the urea-linked prodrugs (compounds 16a and 16c) was more effective, suggesting that urea linked prodrugs were better candidates for MDEPT than thiourea analogs.
The 1-aryl-3,3-dimethyltriazenes are also known antineoplastic drugs, however, this class of compounds exhibited low selectivity for cancer cells. In order to solve this problem, new triazene prodrugs for MDEPT were synthesized by Perry and co-workers [1]. In this work, tyramine (17a-17d) or dopamine (17e-17g) subunits were selected as substrates for tyrosinase (Table 1).
![]()
Table 1: Half-lives (t1/2) of triazene prodrugs (17a-17g) in PBS buffer, 80% human plasma and in the presence of tyrosinase (100 U/mL) at 37°C.
View Table 1
These Prodrugs 17a-17g were very stable using phosphate buffer and human plasma. In addition, they were substrate of the enzyme tyrosinase. All compounds have exhibited short half-lives that ranged from 6.1 min - 18.2 min.
Monteiro et al. [28] obtained new triazene derivatives (18a-18f) for MDEPT with good results (Table 2). The half-lives in the presence of the enzyme ranged from 9 (18d) to 20 min (18a). The compound 18c displayed the high cytotoxicity against three different melanoma cell lines. Moreover, this compound (18c) demonstrated an increased specificity for tyrosinase when compared to temozolomide, a commercially available prodrug with tyrosinase-independent toxicity.
![]()
Table 2: Half-lives (t1/2) of triazene derivatives (18a-18f) in PBS buffer, 80% human plasma and in the presence of tyrosinase (300 U/mL) at 37°C.
View Table 2
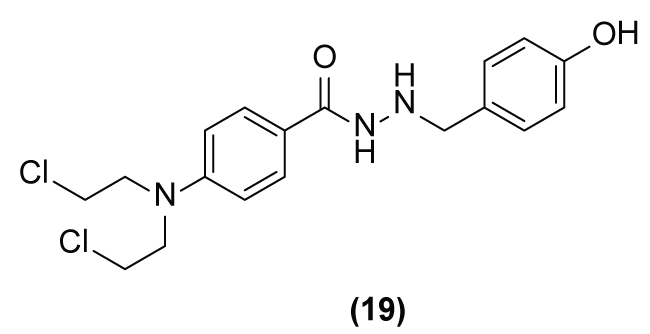
Gasowska-Bajger and Wojtasek [6] have described that not only carbamate or urea linkers, but also hydrazine linkers and phenolic substrates (4-tert-butylcatechol or N-acetyl-L-tyrosine) can be used in drug design for MDEPT. Therefore, they synthetized an aniline mustard prodrug (19) containing a hydrazine linker and a phenolic activator (Figure 8) [29]. The prodrug (N-{4-[bis-(2-chloroethyl)amino]benzoyl}-N-(4-hydroxybenzyl)hydrazine) (19) showed in vivo low specificity for the melanoma cells and weak activity to reduce melanoma solid tumor size compared to melphalan (cytostatic agent) at doses of 100 μg.
.
Figure 8: Chemical structure of (N-{4-[bis-(2-chloroethyl)amino]benzoyl}-N0-(4-hydroxybenzyl)hydrazine) prodrug (19).
View Figure 8
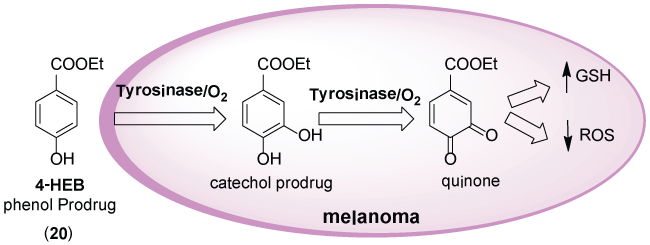
Vad et al. [30] used the same approach to characterize the oxidation and toxicity against five melanocytic melanoma cell lines for 24 phenolic compounds. The phenol prodrug is recognized by the melanoma tyrosinase. After oxidation, it is converted into catechol prodrug that is recognized by tyrosinase being then transformed into aquinone specie. The quinone derivative promotes the decrease of GHS and increase of ROS (Figure 9). The 4-HEB derivative (20) (IC50 = 75 μM) exhibited high selective activity against cell lines, which expressed functional tyrosinase.
.
Figure 9: Mechanism of ethyl 4-hydroxybenzoate (4-HEB) (20) bioconversion by tyrosinase (Adapted from [30]).
View Figure 9
These authors also extended the study for acetaminophen [31] and acetylsalicylic acid [32] and reported that both are tyrosinase substrates. This is an interesting point to be explored in the future for prodrug design. They suggested that the mechanism of toxicity of drugs is due to o-quinone formation, ROS formation, intracellular GSH depletion and mitochondrial toxicity.
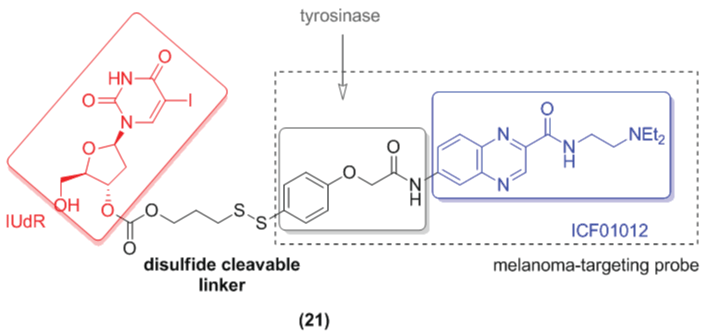
Glutathione (GSH) is also overexpressed in tumor cells at levels 1000times superior to that of the blood plasma [29]. Based on this knowledge, Aissi and co-workers (2015) designed the prodrug (21) based on a melanin-targeting derivative N-(2-diethylaminoethyl)-6-iodoquinoxaline-2-carboxamide (ICF01012). The ICF01012 was conjugated with a self-immolated disulfide linker with the chemotherapeutic IUdR (idoxuridine), an antimetabolite drug. The compound (21) (Figure 10) was able to release IUdR under disulfide reductive conditions. This data suggests that this compound has potential effectiveness to be activated into the tumor cell [33].
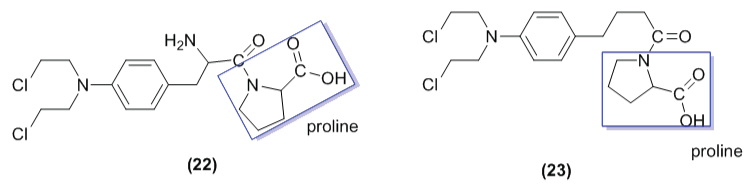
Mittal and co-workers [34] identified using bioinformatics tools that the enzyme prolidase could be an interesting target to development new selective prodrugs for melanoma treatment. The levels of prolidase in malignant cells are high. In addition, this enzyme has high substrate specificity for dipeptides containing proline at the carboxyl terminus [35]. Then, the researchers designed new L- and D- proline prodrugs of melphalan (22a-22b), named as prophalan-L and prophalan-D, respectively (Figure 11).
.
Figure 11: Chemical structure of prophalan, a prodrug of proline and melphalan (22a= L-prophalan, 22b= D-prophalan) and chlorambucil prodrug (23).
View Figure 11
They also reported the bioconversion and antiproliferative activity of these derivatives against melanoma cell line (SK-MEL-5). The results showed that prophalan-L was 7-times more susceptible to the action of prolidase than prophalan-D. The cytotoxic activity of prophalan-L (measured as growth inhibition; GI50 = 74.8 μM) was slightly weaker than that of melphalan (GI50 = 57.0 μM). In this assay, prophalan-D was ineffective, suggesting the necessity of activation of the prodrug by prolidase.
The same research group synthesized the prodrug of chlorambucil (23), mustard structurally similar to melphalan. Unfortunately, this compound exhibited low hydrolysis when incubated with porcine kidney prolidase [5].
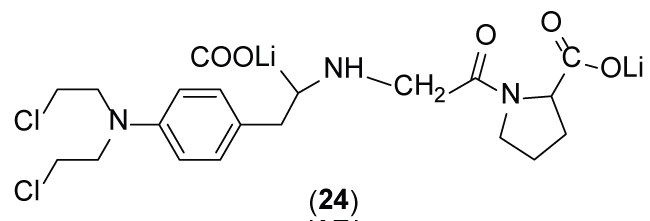
Chrzanowski et al. [36] reported similar coupling of proline moiety to melphalan with few difference in the mode of linkage. In this study, the drug released is the N-substituted melphalan. Despite of the good results, the prodrug (24) (Figure 12) displayed similar susceptibility to the action of prolidase compared to glycyl-proline, an endogenous substrate for prolidase.
.
Figure 12: Chemical structure of prodrug (24) proposed by Chrzanowski et al. (2001).
View Figure 12
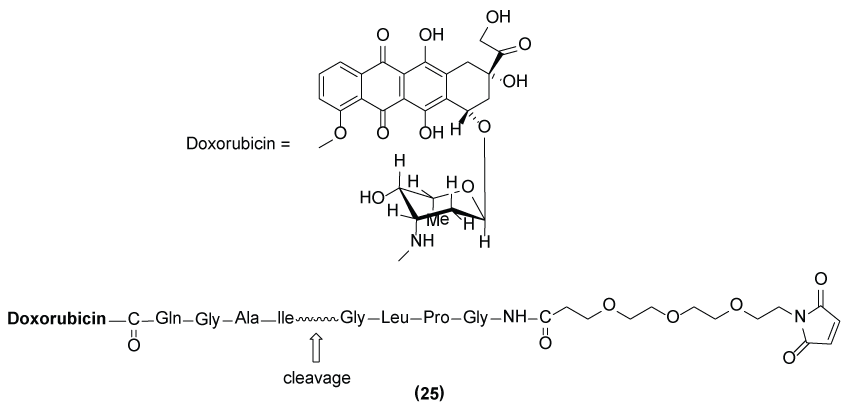
Matrix metalloproteinases (MMPs) are a group of zinc dependent enzymes essential for diverse physiopathological events including regulation of adhesion, migration and growth of cancer cells. MMP-2 and MMP-9 were found to be overexpressed in melanoma tumor cells. This data motivated Mansour and co-workers [37] to explore the protease activity of these enzymes in order to release the antineoplastic agent doxorubicin from a macromolecular carrier (serum albumin) (Figure 13). In vivo data reveals that prodrug (25) demonstrated 4-times superior efficacy than parental drug doxorubicin in mice in the A375 melanoma model.
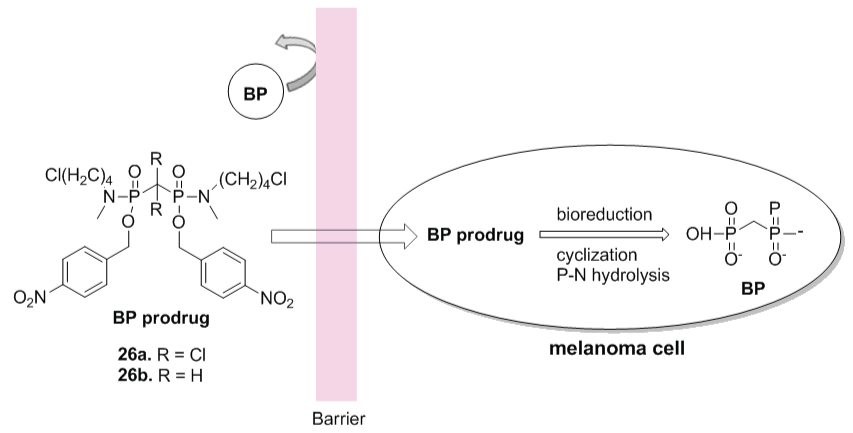
Bisphosphonates are useful to treat osteoporosis and malignant bone disease. These drugs also are inhibitors of adhesion, invasion and growth of cancer cells. Heikkila et al. reported the inhibition of MMP by nitrogen containing bisphosphonates [38]. However, the low cell permeability limits its clinical use. In order to improve the pharmacokinetic profile of these class, Webster et al. [39] synthetized clodronate (26a) and methylene (26b) bisphosphonate prodrugs (Figure 14). All compounds were evaluated against different cancer cells, including melanoma. The compound 26a were able to inhibit different tumor growth cells. This prodrug also displayed selective cytotoxicity against melanoma, with IC50 values of 7 ± 1 μmol/L and 11.7 ± 3 μmol/L at 72 hours against two melanoma cell lines, SK-MEL-5, and UACC-62 respectively.
.
Figure 14: Schematic representation of the bisphosphonate prodrug activation (compounds 26a-26b) (Adapted from [39]).
View Figure 14
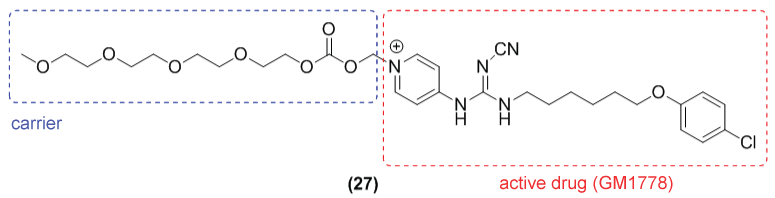
Nicotinamide adenine dinucleotide (NAD) has an important role in several cell functions, including DNA repair [40]. The prodrug (27) GMX1777, Gemin X) (Figure 15) is an inhibitor of the enzyme nicotinamide phosphoribosyl transferase (NAMPT). This enzyme is crucial for the synthesis of NAD+ being overexpressed in cancer cells [41,42]. In previous studies, the compound cyanoguanidinopyridine GMX1778 (active form of the prodrug GMX1777) showed potent anti-tumor activity in several in vivo tumor models as IM-9 (multiple myeloma), HCT-116 (colon carcinoma) and SHP-77 (SCLC) [41,42].
The first results about clinical trial with GMX1777 in patients with advanced malignancies have shown moderate clinical effect after administration of ascending doses (24-hour IV infusion for 21 days). The researches concluded that GMX1777 can be used in combination with other drugs to treat malignant melanoma [43]. Based on these preliminary results, a new phase I/II study of GMX1777 in combination with temozolomide was initiated in patients with metastatic melanoma with a 3-hour infusion for 5 consecutive days every 4 weeks. However, this study was terminated prematurely due to financial constraints [44].
Conclusion
Over the past few decades, the skin cancer is dramatically increasing worldwide. The malignant melanoma is one of the most aggressive melanocytes neoplasm whose treatment is still limited. The current therapy has several limitations that include high toxicity. Therefore, the search of new effective and safe drugs is urgent. Herein, we demonstrated that the prodrug approach is a useful tool to discover new chemotherapeutic agents more selective and less toxic. Metabolic differences between healthy and tumor cells, including the specific enzymes can be explored to get drug selectivity for melanoma. The knowledge of overexpressed enzymes in melanoma tumor such as tyrosinase, prolidase, glutathione, metalloproteases represents an advance in this field that can be explored using prodrug approach. It has been shown that although the studies are still embryonic, the use of targeted prodrug approach point out to clinical success in near future.
Acknowledgements
The authors would like to thank the Brazilian Research Foundation FAPESP Process 2014/08728-6 for financial support.
References
-
Perry MJ, Mendes E, SimplícioAL, CoelhoA, Soares RV, et al. (2009) Dopamine- and tyramine-based derivatives of triazenes: activation by tyrosinase and implications for prodrug design. Eur J Med Chem 44: 3228-3234.
-
Jawaid S, Khan TH, Osborn HM, Williams NA (2009) Tyrosinase activated melanoma prodrugs. Anticancer Agents Med Chem 9: 717-727.
-
Siegel RL, Miller KD, Jemal A (2016) Cancer Statistics, 2016. CA Cancer J Clin 66: 7-30.
-
Foo J, Michor F (2014) Evolution of acquired resistance to anti-cancer therapy. J Theor Biol 355: 10-20.
-
Ge R, Liu L, Dai W, Zhang W, Yang Y, et al. (2016) XPA promotes autophagy to facilitate cisplatin resistance in melanoma cells through the activation of PARP1. J Invest Dermatol.
-
Gasowska-Bajger B, Wojtasek H (2008) Indirect oxidation of the antitumor agent procarbazine by tyrosinase -Possible application in designing anti-melanoma prodrugs. Bioorg Med Chem Lett 18: 3296-3300.
-
Mittal S, Song X, Vig BS, Amidon GL, (2007) Proline Prodrug of Melphalan Targeted to Prolidase, a Prodrug Activating Enzyme Overexpressed in Melanoma. Pharm Res 24: 1290-1298.
-
Batus M, Waheed S, Ruby C, Petersen L, Bines SD, et al. (2013) Optimal management of metastatic melanoma: current strategies and future directions. Am J Clin Dermatol 14: 179-194.
-
Srivastava N, Mc Dermott D (2014) Update on benefit of immunotherapy and targeted therapy in melanoma: the changing landscape. Cancer Manag Res 6: 279-289.
-
Jordan AM, Khan TH, Osborn HM, Photiou A, Riley PA (1999) Melanocyte-directed enzyme prodrug therapy (MDEPT): development of a targeted treatment for malignant melanoma. Bioorg Med Chem 7: 1775-1780.
-
Albert A (1958) Chemical aspects of selective toxicity. Nature 182: 421-422.
-
Chung MC, Silva ATA, Castro LF, Güido RVC, Nassute JC, et al. (2005) Latentiation and advanced drug transport forms. Braz J Pharm Sci 41: 155-179.
-
Mahato R, Tai W, Cheng K (2011) Prodrugs for improving tumor targetability and efficiency. Adv Drug Deliv Rev 63: 659-670.
-
Jornada DH, Dos Santos Fernandes GF, Chiba DE, de Melo TR, Dos Santos JL, et al. (2016) The Prodrug Approach: A Successful Tool for Improving Drug Solubility. Molecules 21: 42.
-
Mandal A, Patel M, Sheng Y, Mitra AK (2015) Design of lipophilic prodrugs to improve drug delivery and efficacy. Curr Drug Targets.
-
Kumar SV, Saravanan D, Kumar B, Jayakumar A (2014) An update on prodrugs from natural products. Asian Pac J Trop Med 7: S54-S59.
-
Ramsay EE, Dilda PJ (2014) Glutathione S-conjugates as prodrugs to target drug-resistant tumors. Front Pharmacol 5: 181.
-
Giang I, Boland EL, Poon GM (2014) Prodrug applications for targeted cancer therapy. AAPS J 16: 899-913.
-
Ettmayer P, Amidon GL, Clement B, Testa B (2004) Lessons learned from marketed and investigational prodrugs. J Med Chem 47: 2393-2404.
-
Miwa M, Ura M, Nishida M, Sawada N, Ishikawa T, et al. (1998) Design of a novel oral fluoropyrimidine carbamate, capecitabine, which generates 5-fluorouracil selectively in tumours by enzymes concentrated in human liver and cancer tissue. Eur J Cancer 34: 1274-1281.
-
Davila L, Ranganathan P (2011) Pharmacogenetics: implications for therapy in rheumatic diseases. Nat Rev Rheumatol 7: 537-550.
-
Zhang J, Stevens MF, Bradshaw TD (2012) Temozolomide: mechanisms of action, repair and resistance. Curr Mol Pharmacol 5: 102-114.
-
Denny BJ, Wheelhouse RT, Stevens MF, Tsang LL, Slack JA (1994) NMR and molecular modeling investigation of the mechanism of activation of the antitumor drug temozolomide and its interaction with DNA. Biochemistry 33: 9045-9051.
-
Tisdale MJ (1987) Antitumor imidazotetrazines--XV. Role of guanine O6 alkylation in the mechanism of cytotoxicity of imidazotetrazinones. Biochem Pharmacol 36: 457-462.
-
Jordan AM, Khan TH, Malkin H, Osborn HM, Photiou A, et al. (2001) Melanocyte-Directed enzyme prodrug therapy (MDEPT): development of second generation prodrugs for targeted treatment of malignant melanoma. Bioorg Med Chem 9: 1549-1558.
-
Seo SY, Sharma VK, Sharma N (2003) Mushroom tyrosinase: Recent prospects. J Agric Food Chem 51: 2837-2853.
-
Knaggs S, Malkin H, Osborn HM, Williams NA, Yaqoob P (2005) New prodrugs derived from 6-aminodopamine and 4-aminophenol as candidates for melanocyte-directed enzyme prodrug therapy (MDEPT) Org Biomol Chem 3: 4002-4010.
-
Monteiro AS, Almeida J, Cabral G, Severino P, Videira PA, et al. (2013) Synthesis and evaluation of N-acylamino acids derivatives of triazenes. Activation by tyrosinase in human melanoma cell lines. Eur J Med Chem 70: 1-9.
-
Frąckowiak-Wojtasek B, Mazurek, Raniszewska, Logghe, Logghe M, et al. (2014) Synthesis and analysis of activity of a potential anti-melanoma prodrug with a hydrazine linker. Eur J Med Chem 71: 98-104.
-
Vad NM, Kandala PK, Srivastava SK, Moridani MY (2010) Structure-toxicity relationship of phenolic analogs as anti-melanoma agents: an enzyme directed prodrug approach. Chem Biol Interact 183: 462-471.
-
Vad NM, Yount G, Moore D, Weidanz J, Moridani MY (2009) Biochemical mechanism of acetaminophen (APAP) induced toxicity in melanoma cell lines. J Pharm Sci 98: 1409-1425.
-
Vad NM, Yount G, Moridani MY (2008) Biochemical mechanism of acetylsalicylic acid (Aspirin) selective toxicity toward melanoma cell lines. Melanoma Res 18: 386-399.
-
El Aissi R, Chezal JM, Tarrit S, Chavignon O, Moreau E (2015) Melanoma-targeted delivery system (part 1): design, synthesis and evaluation of releasable disulfide drug by glutathione. Eur J Med Chem 101: 668-680.
-
Mittal S, Song X, Vig BS, Landowski CP, Kim I, et al. (2005) Prolidase, a potential enzyme target for melanoma: design of proline-containing dipeptide-like prodrugs. Mol Pharm 2: 37-46.
-
Logsdon CD, Simeone DM, Binkley C, Arumugam T, Greenson JK, et al. (2003) Molecular profiling of pancreatic adenocarcinoma and chronic pancreatitis identifies multiple genes differentially regulated in pancreatic cancer. Cancer Res 63: 2649-2657.
-
Chrzanowski K, Bielawska A, Bielawski K, Wołczyński S, Pałka J (2001) Cytotoxicity and effect on collagen biosynthesis of proline analogue of melphalan as a prolidase-convertible prodrug in cultured human skin fibroblasts. Farmaco 56: 701-706.
-
Mansour AM, Drevs J, Esser N, Hamada FM, Badary OA, et al. (2003) A new approach for the treatment of malignant melanoma: enhanced antitumor efficacy of an albumin-binding doxorubicin prodrug that is cleaved by matrix metalloproteinase 2. Cancer Res 63: 4062-4066.
-
Heikkila P, Teronen O, Moianen M, Konttinen YT, Hanemaaijer R, et al. (2002) Bisphosphonates inhibit stromelysin-1 (MMP-3), matrix metalloelastase (MMP-12), collagenase-2 (MMP-13) and enamelysin (MMP-20), but not urokinase-type plasminogen activator, and diminish invasion and migration of human malignant endothelial cells. Anticancer Drugs 13: 245-254.
-
Webster MR, Kamat C, Connis N, Zhao M, Weeraratna AT, et al. (2014) Bisphosphonamidate clodronate prodrug exhibits selective cytotoxic activity against melanoma cell lines. Mol Cancer Ther 13: 297-306.
-
Sampath D, Zabka TS, Misner DL, O'Brien T, Dragovich PS (2015) Inhibition of nicotinamide phosphoribosyltransferase (NAMPT) as a therapeutic strategy in cancer. Pharmacol Ther 151: 16-31.
-
Beauparlant P, Bédard D, Bernier C, Chan H, Gilbert K, et al (2009) Preclinical development of the nicotinamide phosphoribosyl transferase inhibitor prodrug GMX1777. Anticancer Drugs 20: 346-354.
-
Martinsson P, Liminga G, Dhar S, de la Torre M, Lukinius A, et al. (2001) Temporal effects of the novel antitumour pyridyl cyanoguanidine (CHS 828) on human lymphoma cells. Eur J Cancer 37: 260-267.
-
Pishvaian MJ, Marshall JL, Hwang JJ, Malik S, He AR Deeken JF, et al. (2009) A phase I trial of GMX1777, an inhibitor of nicotinamide phosphoribosyl transferase (NAMPRT), given as a 24-hour infusion. J Clin Oncol 27: 15s.
-
Gemin X (2013) A Phase I/II study of GMX1777 in combination with temozolomide for the treatment of metastatic melanoma. ClinicalTrials.gov, NLM Identifier, NCT00724841.
