

International Journal of Diabetes and Clinical Research
Toxic AGEs (TAGE) Theory in the Pathogenesis of NAFLD and ALD
Masayoshi Takeuchi1*, Akiko Sakasai-Sakai1, Jun-ichi Takino2, Takanobu Takata1, Tadashi Ueda1 and Mikihiro Tsutsumi3
1Department of Advanced Medicine, Medical Research Institute, Kanazawa Medical University, Japan
2Laboratory of Biochemistry, Faculty of Pharmaceutical Sciences, Hiroshima International University, Japan
3Department of Hepatology, Kanazawa Medical University, Japan
*Corresponding author:
Masayoshi Takeuchi, Department of Advanced Medicine, Medical Research Institute, Kanazawa Medical University, Uchinada-machi, Ishikawa, 920-0293, Japan, Tel: +81-76-218-8456, Fax: +81-76-286-3652, E-mail: takeuchi@kanazawa-med.ac.jp
Int J Diabetes Clin Res, IJDCR-2-036, (Volume 2, Issue 4), Mini Review; ISSN: 2377-3634
Received: June 25, 2015 | Accepted: July 29, 2015 | Published: July 31, 2015
Citation: Takeuchi M, Sakasai-Sakai A, Takino JI, Takata T, Ueda T, et al. (2015) Toxic AGEs (TAGE) Theory in the Pathogenesis of NAFLD and ALD. Int J Diabetes Clin Res 2:036. 10.23937/2377-3634/1410036
Copyright: © 2015 Takeuchi M, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Nonalcoholic fatty liver disease (NAFLD) and alcoholic liver disease (ALD) have become serious health problems worldwide. These two diseases have similar pathological spectra, ranging from simple hepatic steatosis to steatohepatitis, liver cirrhosis, and hepatocellular carcinoma. NAFLD and ALD are frequently accompanied by extrahepatic complications, including cardiovascular disease and malignancy, which influence patient survival. The chronic ingestion of an excessive daily diet (sweetened beverages and commercial products) increases the levels of the sugar metabolite, glyceraldehyde (GA), while the chronic ingestion of an excessive amount of alcoholic beverages increases the levels of the ethanol metabolite, acetaldehyde (AA) in the liver. GA and AA are known to react non-enzymatically with the amino groups of proteins to form reversible Schiff bases followed by Amadori products. These early glycation products undergo further complex reactions such as rearrangement, dehydration, and condensation to become irreversibly cross-linked, heterogeneous fluorescent derivatives termed "advanced glycation end-products, AGEs" (GA- or AA-AGEs). The Nε-(carboxymethyl)lysine/Nε-(carboxyethyl)lysine or Nε-(ethyl) lysine pathway for the reaction of Amadori products may represent a physiologically relevant mechanism for averting the production of GA- or AA-AGEs and thus, prevent the potential cellular toxicity associated with the formation of GA- or AA-AGEs (toxic AGEs, TAGE) in vivo. The interaction between TAGE and the receptor for AGEs (RAGE) alters intracellular signaling, gene expression and the release of pro-inflammatory molecules and also elicits reactive oxygen species in numerous types of cells, all of which may contribute to the pathological changes observed in lifestyle-related diseases. We herein discussed the pathophysiological roles of GA- and AA-AGEs, the predominant components of TAGE, and a related novel theory for preventing the development and progression of NAFLD and ALD.
Keywords
Metabolic syndrome, Diabetes mellitus, Lifestyle-related diseases
Abbreviations
AGEs: Advanced Glycation End-Products, GA-AGEs: Glycerardehyde-Derived AGEs, AA-AGEs: Acetaldehyde-Derived AGEs, RAGE: Receptor for AGEs, NAFLD: Nonalcoholic Fatty Liver Disease, ALD: Alcoholic Liver Disease
Introduction
Nonalcoholic fatty liver disease (NAFLD) and alcoholic liver disease (ALD) are among the most common causes of chronic liver disease in the westernized world, which now represents a worldwide public health problem [1]. Although these diseases have similar pathological spectra, ranging from simple hepatic steatosis to steatohepatitis and liver cirrhosis [2], many of the characteristics of NAFLD and ALD differ from each other for example, differences in clinical features to patient outcomes. A high total energy intake has been positively associated with the development of NAFLD [3], and specific dietary components have been shown to affect the pathogenesis of this disease. A cross-sectional study reported that a greater intake of soft drinks (SD) was associated with an increased risk of NAFLD [4]. Fructose may also contribute to disease development and progression [5,6]. The increased ingestion of regular SD was recently linked to NAFLD independent of metabolic syndrome [7], with NAFLD patients consuming 5 times more carbohydrates from SD than healthy individuals [8]. The risks of developing and accelerating the progression of ALD are also increased by the intake of alcoholic beverages [9-11]. The types of beverages consumed may also modify the progression of ALD [12]. Drinking patterns are also factors that have been associated with ALD. Previous studies demonstrated that daily or near-daily heavy drinking, not episodic or binge drinking, was closely associated with the development of ALD [9,13]. Moreover, alcohol intake outside of mealtimes and the intake of multiple, different beverages was shown to increase the risk of developing ALD [9].
Advanced glycation end-products (AGEs) are formed by the Maillard reaction, a non-enzymatic reaction between the aldehyde or ketone groups of reducing sugars, such as glucose and fructose and the N-terminal α-amino group or ε-amino group of the lysine residues of proteins, and contribute to the aging of proteins as well as pathological complications associated with diabetes mellitus (DM) [14-21]. In DM-induced hyperglycemia, this process begins with the conversion of reversible Schiff base adducts to more stable, covalently bound Amadori rearrangement products. Over the course of days to weeks, these Amadori products undergo further rearrangement reactions to form irreversibly bound moieties known as AGEs. AGEs were originally characterized by their yellow-brown fluorescent color as well as their ability to form cross-links with and between amino groups; however, this term is now encompasses a broad range of advanced products of the glycation process, including Nε-(carboxymethyl)lysine (CML), Nε-(carboxyethyl)lysine (CEL), and Nε-(ethyl)lysine (NEL), which do not display color or fluorescence and are not cross-linked proteins [14-17]. Recent studies have suggested that AGEs are formed not only from reducing sugars, but also from the carbonyl compounds derived from the auto-oxidation of sugars and other metabolic pathways [17,19-23]. Evidence to show that glyceraldehyde (GA)- and acetaldehyde (AA)-derived AGEs (GA- and AA-AGEs) play a role in the pathogenesis of various disorders, such as DM and diabetic vascular complications, insulin resistance, obesity, hypertension, cardiovascular disease (CVD), Alzheimer's disease (AD), cancer growth/metastasis, and nonalcoholic/alcoholic liver injury, is increasing [18-21,24-31]. In the present study, we discussed the pathophysiological role of GA- and AA-AGEs, predominant components of toxic AGEs (TAGE), and a related novel theory for preventing the development and progression of NAFLD and ALD.
Pathway for the Formation of GA- and AA-AGEs in the Liver
Most people throughout the world consume a combination of two simple sugars, either as table sugar (sucrose) or high-fructose corn syrup (HFCS), both of which are used in many sweetened beverages and commercial products. GA is derived from two distinct pathways in the liver, i) glycolysis and ii) fructolysis [20,21,32]. i) The glycolytic intermediate GA-3-phosphate (G-3-P) is normally catabolized by the enzyme G-3-P dehydrogenase (GAPDH). G-3-P accumulates intracellularly when GAPDH activity decreases. The metabolism of G-3-P then shifts to another route, and the amount of GA is increased. ii) Fructose, a component of sucrose and HFCS, is known to be present in the daily diets of many individuals [33,34]. Fructose is phosphorylated to fructose-1-phosphate by fructokinase and then catabolized to GA and dihydroxyacetone-phosphate by aldolase B [33-36]. Liver damage may occur in patients who consume a daily diet rich in sweetened beverages/commercial products due to the toxicity of GA, which forms GA-AGEs intracellularly and extracellularly (Figure 1).
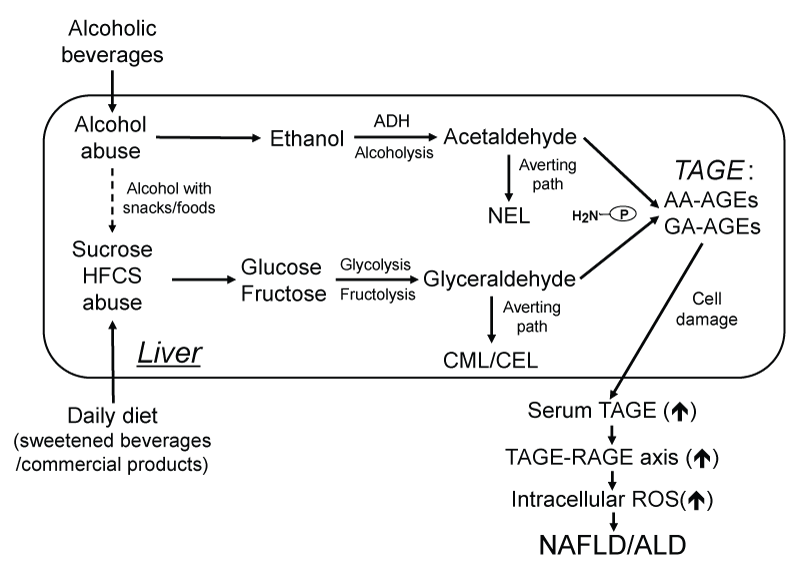
.
Figure 1: Toxic AGEs (TAGE) theory in the pathogenesis of NAFLD and ALD.
The chronic ingestion of an excessive daily diet (sweetened beverages and commercial products) increases the levels of the sugar metabolite, glyceraldehyde (GA), while the chronic ingestion of an excessive amount of alcoholic beverages increases the levels of the ethanol metabolite, acetaldehyde (AA) in the liver. GA and AA are known to react non-enzymatically with the amino groups of proteins to form reversible Schiff bases followed by Amadori products. These early glycation products undergo further complex reactions such as rearrangement, dehydration, and condensation to become irreversibly cross-linked, heterogeneous fluorescent derivatives termed "AGEs" (GA- or AA-AGEs). The CML/CEL or NEL pathway for the reaction of Amadori products may be a physiologically relevant mechanism for averting the production of GA- or AA-AGEs and, thus, prevent the potential cellular toxicity arising from the formation of GA- or AA-AGEs (toxic AGEs, TAGE) in vivo. The interaction between TAGE and the receptor for AGEs (RAGE) has been shown to alter intracellular signaling, gene expression and the release of pro-inflammatory molecules and also elicits reactive oxygen species (ROS) in numerous types of cells, all of which may contribute to the pathological changes observed in lifestyle-related diseases. Taken together, our theory suggests that TAGE are novel therapeutic targets for preventing lifestyle-related diseases. Therefore, inhibiting the formation of TAGE or blocking the TAGE-RAGE axis are promising targets for novel therapeutic interventions against NAFLD/NASH and ALD.
AGEs: Advanced Glycation End-products, AA-AGEs: Acetaldehyde-derived AGEs, ADH: Alcohol Dehydrogenase, ALD: Alcoholic Liver Disease, CEL: Nε-(carboxyethyl)lysine, CML: Nε-(carboxymethyl)lysine, GA-AGEs: Glyceraldehyde-derived AGEs, HFCS: High-Fructose Corn Syrup, NAFLD: Nonalcoholic Fatty Liver Disease, NASH: Nonalcoholic Steatohepatitis, NEL: Nε (ethyl)lysine, RAGE: Receptor for AGEs, ROS: Reactive Oxygen Species, P-NH2: Free Amino Residue of a Protein.
View Figure 1
AA is derived from the alcohol metabolic pathway (alcoholysis). Alcohol is oxidized in the liver, mainly by alcohol dehydrogenase (ADH), to form AA, which is in turn oxidized by aldehyde dehydrogenase (ALDH) to acetate [37]. The cytochrome P450 2E1 (CYP2E1), also contributes to the oxidation of ethanol, but is quantitatively less important than the ADH/ALDH pathway. Both ADH- and CYP2E1-catalyzed oxidation of alcohol generate a reactive metabolite AA, which readily forms adducts with proteins [37]. Liver damage may develop in patients who are abusers of alcohol due to the toxicity of AA, which forms AA-AGEs intracellularly and extracellularly (Figure 1).
GA-AGEs Theory in the Pathogenesis of NAFLD
There is a growing body of evidence to suggest that the interaction between GA-AGEs, but not CML/CEL, and the receptor for AGEs (RAGE) may alter intracellular signaling, gene expression, and the release of pro-inflammatory molecules and also elicits the generation of reactive oxygen species (ROS) in numerous types of cells, all of which may contribute to the pathological changes observed in DM and diabetic vascular complications, hypertension, CVD, AD, and cancer [18-21]. We previously demonstrated that GA-AGEs induced fibrogenesis- and inflammation-related gene and protein expression, such as that of transforming growth factor-β1, collagen type Iα2, and monocyte chemoattractant protein-1, in cultured hepatic stellate cells (HSCs) via the NADPH oxidase-derived generation of ROS [25]. These findings provided a novel insight into how GA-AGEs are involved in the pathogenesis of NAFLD. Furthermore, we found that GA-AGEs induced neurotoxicity and neuronal cell death, whereas CML did not, suggesting that GA-AGEs are also toxic to neuronal cells [38,39]. In AD brains, GA-AGEs were mainly detected in the cytosol of neurons in the hippocampus and parahippocampal gyrus, but not in senile plaques or astrocytes [40]. These findings suggested that the production of GA-AGEs during sugar metabolism may not only be toxic to hepatocytes, but also to neuronal cells and possibly many other cells, thereby inducing cellular and organ impairments.
Regarding the effects of GA-AGEs on hepatocytes, we recently reported that the GA-AGEs-RAGE interaction stimulated hepatic C-reactive protein (CRP) in the human hepatoma cells, Hep3B via the activation of Rac-1 [41]. We demonstrated that GA induced concentration- and time-dependent cell death and increased the intracellular concentration of GA-AGEs in Hep3B cells [42]. We also showed that a GA-AGEs-modified protein of ca.70 kDa, which we identified as heat shock cognate 70, was detected the earliest and in the greatest abundance in GA-treated hepatocytes [42]. We found that intracellular GA-AGEs increased the mRNA expression of the acute phase reactant CRP [42]. These findings prompted us to speculate that extracellular and intracellular GA-AGEs may play roles in the pathogenesis of NAFLD/nonalcoholic steatohepatitis (NASH) (Figure 1).
The excessive intake of fructose contributes to the development of NAFLD and to the progression of the disease to NASH. Fructose is metabolized to GA, which is a precursor of GA-AGEs. We showed that intracellular GA-AGEs were formed in the presence of fructose. Additionally, heterogeneous nuclear ribonucleoprotein M (hnRNPM) was identified as one of the target proteins for GA-AGEs. These findings suggest that GA-modified hnRNPM, resulting from the exposure of the cells to fructose; alter gene expression and causes adverse effects in hepatocytes [43].
The findings of our recent studies revealed that i) the formation of GA-AGEs was enhanced during NASH, and serum and hepatic GA-AGE levels, but not CML, were shown to be significantly higher in patients with NASH than in healthy controls or patients with simple steatosis [24,26], ii) atorvastatin, a hydroxymethyl-glutaryl (HMG)-CoA reductase inhibitor, reduced serum TAGE levels in NASH patients with dyslipidemia [44]. We also recently reported that circulating GA-AGE levels were significantly higher in non-B or non-C hepatocellular carcinoma (HCC) patients than in NASH subjects without HCC or control subjects [45]. These findings suggested that GA-AGEs may play a critical role in the pathogenesis of NASH and may serve as potential targets for therapeutic interventions [46-48].
AA-AGEs Theory in the Pathogenesis of ALD
The pathogenesis of ALD is a dynamic process that is triggered by complex interactions between the metabolic intermediates of alcohol, inflammation, and immune responses from cellular injury [49,50]. Since hepatocytes are the primary site of alcohol detoxification, its major toxic metabolic intermediate, AA causes direct hepatocyte damage and also forms adducts with proteins and DNA [51,52]. The intralobular distribution of AA-AGEs was monitored during the chronic administration of ethanol and also during alcohol abstinence, and was co-evaluated with the progression of ALD and production of ROS [28]. We also demonstrated that the viability of rat primary hepatocytes cultured with AA-AGEs was significantly lower than that of hepatocytes cultured with NEL [28]. Furthermore, we showed that AA-AGEs induced neurotoxicity and neuronal cell death, whereas NEL did not, suggesting that AA-AGEs may also be toxic to neuronal cells [53,54]. We also detected the AA-AGE epitope in the brains of alcoholic individuals [53,54]. These findings suggested that the production of AA-AGEs during alcoholism may not only be toxic to hepatocytes, but also to neuronal cells via RAGE and potentially many other cells, thereby inducing cellular and organ impairments.
The chronic administration of ethanol was shown to result in hepatic fatty degeneration, the severity of which increased between 4 and 8 weeks. We observed a marked increase in staining for AA-AGEs in the pericentral areas of rat livers treated with ethanol [28]. Staining intensity significantly increased from 4 to 8 weeks, indicating that the production of AA-AGEs during the chronic consumption of ethanol was additive and also in parallel with the intensity of hepatic fatty degeneration and ALD. These findings suggested that AA-AGEs play an important role in the production of selective pericentral cytotoxicity in ALD. We demonstrated the intense staining of AA-AGEs surrounding the central vein during the pathogenesis of ALD as well as during the early abstinence period [28]. AA-AGEs that accumulated during the chronic ingestion of ethanol were eliminated with abstinence, during which rats were administered a control liquid diet for a period of 12 weeks following the chronic administration of ethanol for 8 weeks. The intensity of staining for AA-AGEs markedly increased in the hepatocytes of the perivenular region until week 8 during abstinence, but was significantly reduced by 10 weeks and completely disappeared after 12 weeks. Steatosis also simultaneously and completely disappeared after 12 weeks with the restoration of the lobular architecture of hepatic tissue [28]. These findings suggested that the AA-AGEs formed during the chronic consumption of ethanol could be eliminated after abstinence, which helped impaired hepatic tissue to return to its normal function. These findings also led us to speculate that extracellular and intracellular AA-AGEs may play roles in the pathogenesis of ALD.
Furthermore, in vitro cell culture studies using rat HSCs demonstrated that AA-AGEs induced oxidative stress and produced ROS in cultures [28]. HSCs are the main extracellular matrix-producing cells in the liver, and, thus, play a pivotal role in liver fibrogenesis [55]. These findings suggested that AA-AGEs via RAGE induced the production of ROS and contributed to the pathogenesis of ALD. The AA-AGEs thus formed bind to the cellular membrane through RAGE, induce oxidative stress, and produce ROS, which trigger steatosis, hepatocyte ballooning, and the pathogenesis of ALD. These findings led us to speculate that intracellular and extracellular AA-AGEs may play roles in the pathogenesis of ALD (Figure 1).
Conclusion
NAFLD and ALD are generally related to unhealthy lifestyle habits, including the excessive daily intake of sweetened beverages/commercial products and alcohol, and both are likely to be serious health problems in the future. In contrast to chronic viral liver diseases, NAFLD and ALD are frequently accompanied by extrahepatic diseases that can influence patient survival. We demonstrated that GA-AGEs, but not CML, may play a role in the pathogenesis of NASH in humans [24,26,44]. GA-AGEs via RAGE stimulated the proliferation and activation of HSCs in vitro, thereby causing hepatic inflammation and fibrosis [25]. In humans, serum levels of GA-AGEs were significantly higher in NASH patients than in those with simple steatosis and healthy controls [24,26]. We also demonstrated that AA-AGEs were toxic to hepatocytes, whereas NEL was not. The chronic consumption of alcohol produced AA-AGEs in pericentral hepatocytes from the metabolic products of ethanol, mainly AA, and induced oxidative stress, leading to the production of ROS in a rat model. The staining of AA-AGEs was correlated with the severity of ALD in both rats and humans. The chronic ingestion of alcohol has been shown to produce AA-AGEs that contribute to the pathogenesis of ALD [28].
In conclusion, of the various types of AGE structures formed in vivo, the structures of TAGE (GA- and AA-AGEs), but not those of non-TAGE (including CML/CEL and NEL) are likely to play an important role in the pathophysiological processes associated with the formation of AGEs (Figure 1) [17,19-21,28,46,48,53].
Acknowledgments
This work was supported in part by JSPS KAKENHI Grant Number 19300254, 22300264, & 25282029 (for Takeuchi), by MEXT: Regional Innovation Strategy Support Program (for Takeuchi), and by Grant No. SR2012-04 (for Tsutsumi) for research from Kanazawa Medical University.
References
-
Vuppalanchi R, Chalasani N (2009) Nonalcoholic fatty liver disease and nonalcoholic steatohepatitis: Selected practical issues in their evaluation and management. Hepatology 49: 306-317.
-
Tannapfel A, Denk H, Dienes HP, Langner C, Schirmacher P, et al. (2011) Histopathological diagnosis of non-alcoholic and alcoholic fatty liver disease. Virchows Arch 458: 511-523.
-
Sullivan S (2010) Implications of diet on nonalcoholic fatty liver disease. Curr Opin Gastroenterol 26: 160-164.
-
Zelber-Sagi S, Nitzan-Kaluski D, Goldsmith R, Webb M, Blendis L, et al. (2007) Long term nutritional intake and the risk for non-alcoholic fatty liver disease (NAFLD): a population based study. J Hepatol 47: 711-717.
-
Ouyang X, Cirillo P, Sautin Y, McCall S, Bruchette JL, et al. (2008) Fructose consumption as a risk factor for non-alcoholic fatty liver disease. J Hepatol 48: 993-999.
-
Abdelmalek MF, Suzuki A, Guy C, Unalp-Arida A, Colvin R, et al. (2010) Increased fructose consumption is associated with fibrosis severity in patients with nonalcoholic fatty liver disease. Hepatology 51: 1961-1971.
-
Assy N, Nasser G, Kamayse I, Nseir W, Beniashvili Z, et al. (2008) Soft drink consumption linked with fatty liver in the absence of traditional risk factors. Can J Gastroenterol 22: 811-816.
-
Abid A, Taha O, Nseir W, Farah R, Grosovski M, et al. (2009) Soft drink consumption is associated with fatty liver disease independent of metabolic syndrome. J Hepatol 51: 918-924.
-
Bellentani S, Saccoccio G, Costa G, Tiribelli C, Manenti F, et al. (1997) Drinking habits as cofactors of risk for alcohol induced liver damage The Dionysos Study Group. Gut 41: 845-850.
-
Becker U, Deis A, Sorensen TI, Gronbaek M, Borch-Johnsen K, et al. (1996) Prediction of risk of liver disease by alcohol intake, sex, and age: a prospective population study. Hepatology 23: 1025-1029.
-
Naveau S, Giraud V, Borotto E, Aubert A, Capron F, et al. (1997) Excess weight risk factor for alcoholic liver disease. Hepatology 25: 108-111.
-
Kerr WC, Fillmore KM, Marvy P (2000) Beverage-specific alcohol consumption and cirrhosis mortality in a group of English-speaking beer-drinking countries. Addiction 95: 339-346.
-
Hatton J, Burton A, Nash H, Munn E, Burgoyne L, et al. (2009) Drinking patterns, dependency and life-time drinking history in alcohol-related liver disease. Addiction 104: 587-592.
-
Bucala R, Cerami A (1992) Advanced glycosylation: chemistry, biology, and implications for diabetes and aging. Adv Pharmacol 23: 1-34.
-
Vlassara H, Bucala R, Striker L (1994) Pathogenic effects of advanced glycosylation: biochemical, biologic, and clinical implications for diabetes and aging. Lab Invest 70: 138-151.
-
Brownlee M (1995) Advanced protein glycosylation in diabetes and aging. Ann Rev Med 46: 223-234.
-
Takeuchi M, Makita Z (2001) Alternative routes for the formation of immunochemically distinct advanced glycation end-products in vivo. Curr Mol Med 1: 305-315.
-
Takeuchi M, Yamagishi S (2004) TAGE (toxic AGEs) hypothesis in various chronic diseases. Med Hypotheses 63: 449-452.
-
Sato T, Iwaki M, Shimogaito N, Wu X, Yamagishi S, et al. (2006) TAGE (toxic AGEs) theory in diabetic complications. Curr Mol Med 6: 351-358.
-
Takeuchi M, Yamagishi S (2009) Involvement of toxic AGEs (TAGE) in the pathogenesis of diabetic vascular complications and Alzheimer's Disease. J Alzheimer Dis 16: 845-858.
-
Takeuchi M, Takino J, Yamagishi S (2010) Involvement of the toxic AGEs (TAGE)-RAGE system in the pathogenesis of diabetic vascular complications: A novel therapeutic strategy. Curr Drug Targets 11: 1468-1482.
-
Glomb MA, Monnier VM (1995) Mechanism of protein modification by glyoxal and glycolaldehyde, reactive intermediates of the Maillard reaction. J Biol Chem 270: 10017-10026.
-
Thornalley PJ, Langborg A, Minhas HS (1999) Formation of glyoxal, methylglyoxal and 3-deoxyglucosone in the glycation of proteins by glucose. Biochem J 344: 109-116.
-
Hyogo H, Yamagishi S, Iwamoto K, Arihiro K, Takeuchi M, et al. (2007) Elevated levels of serum advanced glycation end products in patients with non-alcoholic steatohepatitis. J Gastroenterol Hepatol 22: 1112-1119.
-
Iwamoto K, Kanno K, Hyogo H, Yamagishi S, Takeuchi M, et al. (2008) Advanced glycation end products enhance the proliferation and activation of hepatic stellate cells. J Gastroenterol 43: 298-304.
-
Hyogo H, Yamagishi S (2008) Advanced glycation end products (AGEs) and their involvement in liver disease. Curr Pharm Des 14: 969-972.
-
Yamagishi S, Maeda S, Matsui T, Ueda S, Fukami K, et al. (2012) Role of advanced glycation end products (AGEs) and oxidative stress in vascular complications in diabetes. Biochim Biophys Acta 1820: 663-671.
-
Hayashi N, George J, Takeuchi M, Fukumura A, Toshikuni N, et al. (2013) Acetaldehyde-derived advanced glycation end-products promote alcoholic liver disease. PLoS One 8: e70034.
-
Takino J, Yamagishi S, Takeuchi M (2012) Glycer-AGEs-RAGE signaling enhances the angiogenic potential of hepatocellular carcinoma by upregulating VEGF expression. World J Gastroenterol 18: 1781-1788.
-
Takeuchi M, Yamagishi S (2008) Possible involvement of advanced glycation end-products (AGEs) in the pathogenesis of Alzheimer's disease. Curr Pharm Des 14: 973-978.
-
Abe R, Yamagishi S (2008) AGE-RAGE system and carcinogenesis. Curr Pharm Des 14: 940-945.
-
Takeuchi M, Yamagishi S (2004) Alternative routes for the formation of glyceraldehyde-derived AGEs (TAGE) in vivo. Med Hypotheses 63: 453-455.
-
Schalkwijk CG, Stehouwer CD, Van Hinsbergh VW (2004) Fructose-mediated non-enzymatic glycation: sweet coupling or bad modification. Diabetes Metab Res Rev 20: 369-382.
-
Gaby AR (2005) Adverse effects of dietary fructose. Altern Med Rev 10: 294-306.
-
Hallfrisch J (1990) Metabolic effects of dietary fructose. FASEB J 4: 2652-2660.
-
Mayes PA (1993) Intermediary metabolism of fructose. Am J Clin Nutr 58: 754S-765S.
-
Lieber CS (2004) Alcoholic fatty liver: its pathogenesis and mechanism of progression to inflammation and fibrosis. Alcohol 34: 9-19.
-
Takeuchi M, Bucala R, Suzuki T, Ohkubo T, Yamazaki M, et al. (2000) Neurotoxicity of advanced glycation end-products for cultured cortical neurons. J Neuropathol Exp Neurol 59: 1094-1105.
-
Takeuchi M, Kikuchi S, Sasaki N, Suzuki T, Watai T, et al. (2004) Involvement of advanced glycation end-products in Alzheimer's disease. Curr Alzheimer Res 1: 39-46.
-
Choei H, Sasaki N, Takeuchi M, Yoshida T, Ukai W, et al. (2004) Glyceraldehyde-derived advanced glycation end products in Alzheimer's disease. Acta Neuropathol 108: 189-193.
-
Yoshida T, Yamagishi S, Nakamura K, Matsui T, Imaizumi T, et al. (2006) Pigment epithelium-derived factor (PEDF) inhibits advanced glycation end product (AGE)-induced C-reactive protein expression in hepatoma cells by suppressing Rac-1 activation. FEBS Lett 580: 2788-2796.
-
Takino J, Kobayashi Y, Takeuchi M (2010) The formation of intracellular glyceraldehyde-derived advanced glycation end-products and cytotoxicity. J Gastroenterol 45: 646-655.
-
Takino J, Nagamine K, Takeuchi M, Hori T (2015) In vitro identification of nonalcoholic fatty liver disease-related protein hnRNPM. World J Gastroenterol 21: 1784-1793.
-
Kimura Y, Hyogo H, Yamagishi S, Takeuchi M, Ishitobi T, et al. (2010) Atorvastatin decreases serum levels of advanced glycation endproducts (AGEs) in nonalcoholic steatohepatitis (NASH) patients with dyslipidemia: clinical usefulness of AGEs as a biomarker for the attenuation of NASH. J Gastroenterol 45: 750-757.
-
Kan H, Yamagishi S, Ojima A, Fukami K, Ueda S, et al. (2014) Elevation of serum levels of advanced glycation end products in patients with non-B or non-C hepatocellular carcinoma. J Clin Lab Anal.
-
Takeuchi M, Takino J, Sakasai-Sakai A, Takata T, Ueda T, et al. (2014) Involvement of the TAGE-RAGE system in NASH: Novel treatment strategies. World J Hepatol 6: 880-893.
-
Takeuchi M, Sakasai-Sakai A, Takata T, Ueda T, Takino J, et al. (2015) Serum levels of toxic AGEs (TAGE) may be a promising novel biomarker in development and progression of NASH. Med Hypotheses 85: 490-493.
-
Takino J, Nagamine K, Hori T, Sakasai-Sakai A, Takeuchi M (2015) Contribution of the toxic AGEs (TAGE)-RAGE axis in NASH-related HCC. World J Gastroenterol.
-
Beier JI, McClain CJ (2010) Mechanisms and cell signaling in alcoholic liver disease. Biol Chem 391: 1249-1264.
-
Gao B, Bataller R (2011) Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology 141: 1572-1585.
-
Setshedi M, Wands JR, Monte SM (2010) Acetaldehyde adducts in alcoholic liver disease. Oxid Med Cell Longev 3: 178-185.
-
Yu HS, Oyama T, Isse T, Kitagawa K, Pharm TT, et al. (2010) Formation of acetaldehyde-derived DNA adducts due to alcohol exposure. Chem Biol Interact 188: 3672-3675.
-
Takeuchi M, Watai T, Sasaki N, Choei H, Iwaki M, et al. (2003) Neurotoxicity of acetaldehyde-derived advanced glycation end products for cultured cortical neurons. J Neuropathol Exp Neurol 62: 486-496.
-
Takeuchi M, Saito T (2005) Cytotoxicity of acetaldehyde-derived advanced glycation end-products (AA-AGE) in alcoholic-induced neuronal degeneration. Alcohol Clin Exp Res 29(12 Suppl): 220S-224S.
-
Moreira RK (2007) Hepatic stellate cells and liver fibrosis. Arch Pathol Lab Med 131: 1728-1734.
