

International Journal of Diabetes and Clinical Research
The Crossroad of Inflammation and Diabetes: Role of Toll-Like Receptor
Behrouz Salehian1* and Vahid Mahabadi2
1Department of Clinical Diabetes, Endocrinology and Metabolism, City of Hope Diabetes & Metabolism Research Institute, USA
2David Geffen School of medicine, University of California, Los Angeles, USA
*Corresponding author:
Behrouz Salehian, Department of Clinical Diabetes, Endocrinology and Metabolism, City of Hope Diabetes & Metabolism Research Institute, Los Angeles, USA, bsalehian@coh.org
Int J Diabetes Clin Res, IJDCR-2-032, (Volume 2, Issue 3), Review Article; ISSN: 2377-3634
Received: April 08, 2015 | Accepted: June 27, 2015 | Published: June 29, 2015
Citation: Salehian B, Mahabadi V (2015) The Crossroad of Inflammation and Diabetes: Role of Toll-Like Receptor. Int J Diabetes Clin Res 2:032. 10.23937/2377-3634/1410032
Copyright: © 2015 Salehian B, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
A different facet of type 2 diabetes was recognized in the early 80s with the demonstration of the "state of coagulation activation" and "endothelial dysfunction / activation" in not only type 2 diabetes but also in obesity. The discovery of tumor necrosis factor (TNF) alpha and its physiological role including many aspects of coagulation activation and endothelial dysfunction was later found to have a correlation with adiposity and type 2 diabetes. Further knowledge in the metabolic function of TNF and concomitant discovery of elevated cytokines released from adipose tissue suggested the integral involvement of inflammation in the pathogenesis of type 2 diabetes and obesity. The increased influx of monocytes in adipose tissue, an inflammatory state, elevates TNF alpha secretion leading to the induction of insulin resistance through the release of free fatty acids from visceral adipose tissue; furthermore, TNF alpha directly causes insulin receptor signaling dysfunction by shifting the phosphorylation residues.
The fundamental question of why excess adiposity stimulates inflammatory response, thereby insulin resistance, lingered for a decade. The demonstration of free fatty acids (lauric, palmitic and stearic acid) engaging Toll Like Receptor 4 to stimulate innate immunity has some similarity to bacterial lipopolysacharides; providing the link of visceral adiposity to inflammatory syndrome.
This article provides a historical review of the progression of knowledge in unmasking the inflammatory aspect in diabetes and visceral obesity, which were diseases often associated with hyperglycemia. In fact, it is the inflammatory aspect of the disease that governs the basic pathophysiology with complex interplays at the cross road of inflammation and metabolism.
Keywords
Diabetes, Inflammation, Obesity, Metabolism, Toll-like-receptor, Tumor Necrosis Factor Alpha, Monocyte
Abbreviations
11BHSD1: 11 Beta Hydroxysteroid Dehydrogenase Type 1, AMPK: AMP Kinase, FFA: Free Fatty Acids, ICAM-1: Intercellular Adhesion Molecule-1, IL: Interleukin, IRS-1: Insulin Receptor Substrate-1, LPL: Lipoprotein Lipase, MAP: Mitogen Activated Protein, MCP-1: Monocyte-Derived Chemotactic Protein -1, PAI 1: Plasminogen Activator Inhibitor Type 1, PI3K: Phosphatydil Inositol 3 Kinase, TAT: Thrombin-Antithrombin Complex, tPa: Tissue Plasmiogen Activator, TLR: Toll-Like Receptor, TNF: Tumor Necrosis Factor, VCAM-1: Vascular-Cell Adhesion Molecule-1, VWF: Von Willebrand Factor
Introduction
Part 1- Early Findings in Type 2 Diabetes: How the shift was made from a metabolic disease to an inflammatory disease
Coagulation Activation and Endothelial Dysfunction in Type 2 Diabetes Mellitus
Since the mid-80s, it has become progressively accepted that patients with type 2 diabetes are in procoagulant state with elevated Tissue Factor in their plasma, which has been suggested to activate the coagulation cascade. Increased thrombin activity as measured by fibrinogen peptide derivative B beta [1], or Thrombin-antithrombin complex (TAT), points to a constant and latent activation of coagulation, and an inability of endothelium to defend itself by adequately releasing tissue plasminogen activator (tPa) in type 2 diabetic patients. In one study, it has been shown that the state of activation of coagulation are promoting clot formation in person with diabetes and stable angina, as oppose to non diabetic, non-obese person with stable angina [2]. Furthermore the tpA release from the endothelium, one of the last line of defense against formed clots, is abnormally absent or low in diabetic patients with angina [2] or without angina [3]. Other elements of coagulation activation in type 2 diabetes are fibrin degradation product, factor VIIa, thrombin activation product and fibrinogen [4]. The procoagulant state in type 2 diabetes is unrelated to the presence or absence of complications [4]. In particular, plasma plasminogen activator inhibitor type 1 (PAI 1) is constantly elevated among patients with type 2 diabetes [5], whereas the markers of fibrinolysis are insufficient, and the balance usually tilt towards procoagulant state.
Use of invasive and non-invasive methods demonstrated an inability of arterial vasodilatory response when exposed to ischemia in patients with type 2 diabetes. As a prelude to an extensive examination of endothelium as a culprit to abnormal coagulant response in diabetic patient; earlier work clearly demonstrated von Willebrand factor (VWF) as an endothelial distress signal [5] and E-selectin as an index of endothelial activation released from endothelium (and promoting the activation of platelets) in diabetic patient with coronary artery disease [6]. Interestingly, better glycemic control has no impact on improving endothelial activation and procoagulant state [7]. However, in a cross-sectional study, the levels of thrombin generation after platelet activation with calcium chloride tend to be associated with the degree of glycemic control, where subjects with poor glycemic control (HbA1c ≥9) were associated with higher thrombin generation and shorter clotting time [8] in comparison to the control and subjects with good glycemic control (HbA1c <9). It has been shown in another study that improvement of glycemic control with troglitazone, a first drug of the thiazolodinedione, was associated with decrease thrombogenicity [9] as measured by clot generation using Badimon perfusion chamber to mimic abnormal rheology of coronary arteries. Troglitazone, a PPAR gamma agnonist is known to have anti-inflammatory effect through adiponectin synthesis [10] and decreases TNF alpha level [11]. It is unknown whether this antithrombogenic effect is mediated by improvement of glycemic control or the reduction in inflammatory cytokines. E-Selectin represents activation of endothelium, an event that usually takes place with inflammatory response. As a marker of endothelial activation, E-selectin correlates with visceral obesity, while glycemic control has no or minimal effect. Waist to hip ratio has been shown to be correlated to E-selectin, and interestingly, weight reduction causes E-selectin reduction suggesting the process of endothelial dysfunction and procoagulant state is related to adiposity and inflammation but not as the result of hyperglycemia per se in type 2 diabetics [12]. Furthermore, elevated levels of intercellular adhesion molecule-1 (ICAM-1), vascular-cell adhesion molecule-1 (VCAM-1), P-selectin and major cytokines (TNF alpha and IL-6) were found in obese women and is associated with visceral obesity. Conversely, weight loss leads to decrease in the serum level of cytokines (TNF alpha and IL-6), E- selectin, and normalization of endothelial response to vasodilators [13]. Thus, not only obesity is associated with activation of coagulation, but also the involvement of cytokine signaling, while the role of hyperglycemia seems absent in this complex interaction. Both the activation of coagulation and elevation of pro-inflammatory cytokines level are interconnected with visceral adiposity; therefore, endothelial dysfunction and coagulation activation poses a greater risk for a diabetic obese patient for cardiovascular events.
Cytokines and Coagulation Activation
Cachectin is a hormone secreted by macrophages with the ability to suppress lipoprotein lipase (LPL) activity in 3T3-1 adipose cell line [14]. This hormone later became widely known as tumor necrosis factor (TNF) Alpha [15,16] and has been shown as a prototype of inflammatory cytokines to promote tissue factor activation, and promoting coagulation pathway activation. Tissue factor is believed to be a major initiator of coagulation in vivo. TNF alpha concomitantly decreases activation of protein C, and increase the release of protein S from endothelium. During active inflammation, thrombin mediated protein C activation and formation of functional activated protein C-and Protein S complex on the endothelial lining were attenuated, promoting shift toward coagulation, while natural anticoagulants (protein C and S) are ineffective to protect endothelium [16]. Interestingly, both TNF and Interleukin(IL)1 were found to increase the level of PAI-1 and decrease the tPA release from endothelium in a dose dependent manner [17]; a condition that was frequently observed not only in type 2 diabetes mellitus but also in visceral obesity and further demonstrates the improvement in endothelial fibrinolytic reaction due to attenuation in inflammatory syndrome. While the role of TNF alpha released from macrophage as a response of host to the lipoproteins of bacterial cell wall in septic shock was well known in late-80s [18], some 20 years later, TNF alpha is providing the link between obesity and diabetes, with the discovery of Toll Like Receptors (TLR).
Cytokines and Endothelial Activation
Tumor Necrosis Factor alpha as described has been shown to activate endothelial cells in vitro and promote platelet thrombus along with fibrinopeptide A and fibrin deposition on endothelial surface [19], which led to the discovery of vascular permeability factor induced by cytokines in mouse bearing TNF sensitive tumor. This vascular permeability factor was found to have the ability to activate macrophages and monocytes with chemotactic effect on crossing the endothelium [20], which plays a major role in atherosclerosis. The effect of TNF on endothelium is mediated mostly through the P55 endothelial surface receptor for expressing cell adhesion molecules, mediating macrophages migration in atherosclerosis and accentuating inflammatory response in and around endothelium e.g. smooth muscle cells [21].
Interestingly, similar changes and endothelial activation was found to be induced by advanced glycated byproducts, glycated proteins that accumulate in rapid rate in the vasculature of diabetic patient [22]. Treatment of poorly controlled diabetic patients in a randomized cross-over study using insulin and sulfonylurea over a course of 16 weeks each did not attenuate the increased levels of markers of activation of endothelium, coagulation and inflammation despite improved glycemic control [23]. It was already known that many aspects of endothelial activation and state of inflammation found in diabetes has a counterpart in patient with obesity. Similar to diabetic person, obese person who has visceral fat, not only has similar dyslipidemia as a person with diabetes/ metabolic syndrome, but also exhibit increase in tissue factor VII, elevated PAI -1, endothelial dysfunction, along with features of inflammatory syndrome including IL-6 levels, C- reactive protein [24]. The similarity of endothelial response in inflammatory condition with visceral obesity and type 2 diabetes led to the concept of inflammation induced insulin resistance [25].
Inflammation and Adipocytes
One of the characteristic effects of TNF recognized early on was the suppression of LPL synthesis in 3T3-L1 adipocytes cell line, which was later found to be an oversimplification. The synthesis of the enzyme and its activity actually increases in the liver and overall in the blood of animals (mouse, rats and guinea pigs) [26]. After one single TNF infusion, the adipocytes' LPL synthesis and activity were suppressed for 48 hours and resumed after 1 week, while the adipocytes LPL activity and synthesis declines [26]. However in human adipocytes, TNF alpha has no effect on LPL synthesis and activity [27]. TNF has been shown to increase lipid production, triglycerides, liver LDL production, and hepatic sterol synthesis in humans [28]. Obesity in humans is associated with alteration of metabolic function of adipose tissue with release of free fatty acids, cytokines and change in certain hormones such as resistin and adiponectin levels. Adipose tissue of obese subjects compared to lean subjects has increase expression of TNF alpha, IL-6, iNOS, TGFB, C-reactive protein [29] monocyte-derived chemotactic protein -1(MCP-1) [30]. MCP-1 has been shown by itself affecting the function of adipocytes, decreasing insulin stimulated glucose uptake and expression of several adipogenic genes including lipoprotein lipase, adipsin, glucose transporter-4, beta 3 adrenergic receptors, and most importantly, PPAR Gamma expression [30]. The interaction of TNF and monocytes is self-perpetuating, causing MCP-1 release and further deterioration of glucose uptake and release of free fatty acids. Furthermore, hyperinsulinemia, which is the hallmark and companion of visceral obesity, also stimulates secretion of MCP-1 [30]. MCP-1 is an insulin sensitive hormone, similar to procoagulant factor, PAI -1[30]. The numbers of monocytes and macrophages derived from bone marrow in the adipose tissue positively correlated with adipocyte size and body mass; furthermore, adipose tissue macrophages accounts for almost all of the adipose tissue TNF alpha, IL-6 and iNos expression [31].
In fact, the inflammation that ensues from accumulation of fat is dramatically causing elevation of serum level of insulin in mouse and in humans, initiating the process of diabetogenesis [32]. In genetic mouse model of obesity( db/db, ob/ob) and in fat induced obesity, upregulation of genes and macrophage specific genes such as MIP-1 alpha (macrophage inflammatory protein 1 alpha), ADAM8 (a disintegrin like metalloproteinase), MCP-1, and macrophage antigen -1 (CD 11b) in white adipose tissue precedes the migration of macrophages to adipose tissue, lipolysis and giant cell formation, and dramatic increase in serum insulin levels [32]. The expression of inflammatory genes are restricted or enriched in macrophages [32]. MCP-1 expression is also increased in isolated adipocytes from obese subjects and is associated with insulin resistance. Weight loss is accompanied with not only decrease in serum insulin levels but also the serum levels of MCP-1 [33].
Interleukin 6 has a pleotropic inflammatory response and increases acute phase reactant proteins from the liver; however, it also has anti-inflammtory effects as well [34]. IL-6 secretion during inflammatory process is an association within the inflammatory response of the host, but in fact it is key in controlling the downward spiral of the inflammatory response. It has been shown that weight loss is associated with a decrease in expression of TNF alpha, IL-1 beta, IL-1Receptor and IL-18 in peripheral mononuclear cells, however compared to the control group without weight loss, an increase in expression of IL-6 was observed among those who are loosing weight and sustaining their weight loss [35]. Interestingly, there is a negative correlation between peripheral blood mononuclear cells (PBMC) [35] expression of IL-6 and of insulin levels (tertiles) among subjects with sustained weight loss. This demonstration further highlights the inflammatory components in obesity, and improvements in glycemic control and insulin resistance can be achieved by reducing proinflammatory cytokines and increasing anti-inflammatory cytokines such as IL-6. In another study among obese subjects, increased in serum IL-6 was observed [36]. IL-6 has been demonstrated to improve insulin sensitivity when infused to human volunteers by increasing glucose disposal without affecting endogenous glucose production [37]. IL-6 increases fatty acid oxidation, increases glucose transporter 4 translocation and thereby increases insulin mediated glucose uptake, all through AMP Kinase (AMPK) pathway [37].
Adipokines
Not only inflammatory cytokines such as TNF alpha, IL-6 and MCP-1 are secreted from adipocytes but also a plethora of other hormones. Leptin, the first adipocyte-derived hormone to be discovered, is a major hormone affecting resting energy expenditure and satiety. Next, adiponectin was discovered as a major insulin sensitizing and anti-inflammatory hormone in humans, while resistin was found to be acting against adiponectin. Thus, adipose tissues are engaged in complex interactions to protect insulin sensitivity, when the system is driving the metabolic system toward insulin resistance, diabetogenesis and promotion of atherosclerosis.
Adiponectin
Adiponectin, found abundantly in plasma of healthy humans, is an exclusive adipocyte-derived cytokine, which is composed of a long polypeptide with 244 amino acid (30-kDa), initially characterized by Scherer PE et al. in 1995 [38-40]. The Adiponectin gene is located on chromosome 3q27 [41], which has been linked to metabolic syndrome, type 2 diabetes, anti- atherosclerosis and plays an immunomodulatory role as well (Figure 1).
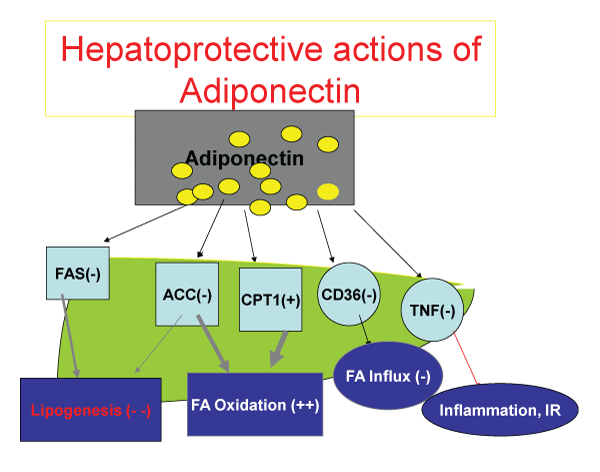
.
Figure 1: The metabolic function of adiponectin. After release from adipoblasts and adipocytes, adiponectin inhibits Fatty-Acyl synthesis both in the liver and muscle, inhibits Acetyl Co-A carboxykinase, while stimulating Carnitine palmitoyl transferase 1 to promote transfer of fatty acid to mitochondria for beta oxidation. The end result is decrease in fatty acid synthesis, promotion of fatty acid oxidation. Adiponectin, by down regulating CD36 receptor, decreases Fatty acid influx to hepatocytes and myocytes, and by inhibition of TNF alpha release and action, decreases inflammatory influence of TNF alpha on free fatty acid release to the circulation and attenuates the insulin resistance.
View Figure 1
Adiponectin is produced in two structurally distinct forms in plasma, which is globular and full-length with 3 species of multimers that are all biologically active. Adiponectin exerts its effect by binding to three receptors discovered so far (AdipoR1, AdipoR2, and T-cadherin-CDH13), with distinct tissue specialties and affinities. In some studies, the cut-off value (in microgram/ml) of adiponectin has been used to determine the development of metabolic syndrome and diabetes mellitus, although it's not fully functional and not a standard for routine practice. Kadowaki et al. showed that hypoadiponectinemia is caused by interactions between both genetic factors such as SNPs in adiponectin gene and environmental factors such as sedentary lifestyle, which leads to obesity [42].
Many observational studies showed beneficial effects of adiponectin in fuel metabolism. Its production by adipocytes directly affects insulin resistance, insulin level, lipid metabolism, and inflammatory responses that are involved in many different pathways by diminishing TLR-4 signaling as a culprit in this process, which will be discussed in more details below.
There is a strong association between adiponectin and the risk of developing type 2 diabetes. Studies showed that in subjects with insulin resistance and hyperinsulinemia, the level of adiponectin is considerably lower. Lower adiponectin level plays a more crucial role in the development of diabetes even when compared to the degree of adiposity and glucose intolerance [43].
Tsuchida et al. showed that the expressions of both adiponectin receptors (AdipoR1 & AdipoR2) are significantly decreased in the muscle and adipose tissue of insulin resistant ob/ob mice, which could be due to obesity-linked hyperinsulinemia [44].
Beside diabetes, adiponectin can exert its anti-inflammatory effects and protection in other disease models. Yamaguchi et al. investigated the potent inhibitory action of adiponectin in inflammatory response and its negative regulation on macrophage-like cell response toward toll-like receptors. They demonstrated the regulatory functions of adiponectin as anti-inflammatory molecule in bacterial and viral infections [45]. Along the same line, Jenke et al. demonstrated in an experimental autoimmune myocarditis mouse model, adiponectin protects against interaction of cardiac and immune cells by diminishing the TLR4 signaling and attenuation of inflammatory response [46]. This work showed adiponectin mediates cardioprotection by modulation of inflammatory response and decreasing the cardiac injury following ischemia. Systemic overexpression of adiponectin resulted in reduction of cardiac expression of TLR4, immune cell infiltration, myocardial injury, NF-κB (which controls pro-inflammatory targets such as TNF-α, IL-6, ICAM-1 & CCL-2), activation and migration of immune cells, and subsequently cardiomyocyte apoptosis [46].
In addition, it has been shown that adiponectin is involved in anti-inflammatory pathways in alcohol liver disease and serves a protective role. Working models showed that chronic alcohol-dependency causes sensitization of TLRs with subsequent cytokine production (TNF-α) by Kupffer cells and liver injury. Adiponectin exerts it anti-inflammatory action through adiponectin receptors on Kupffer cells in the liver [47].
Leptin
Leptin from Greek leptos, which means thin, is a hormone produced by adipose tissue with a crucial role in energy homeostasis in central and peripheral tissues mainly on weight and appetite control. Leptin is the first multifunctional adipose-derived cytokine discovered in 1994 and the gene is located on chromosome 7 in humans and called ob (Lep) gene. The gene codes for production of this polypeptide hormone, which consists of 167 amino acids. Its effect on body exerts through both leptin itself and its receptor (LEPR), where deficiency of either one could cause profound hyperphagia, obesity and strongly correlated with body mass index (BMI) and body fat. Leptin serum concentration reflects the proportion of adipose tissue [48,49]. Subsequent studies demonstrated that leptin is involved in other pathological conditions including cardiovascular disease, immune system, reproductive system, and most recently recognized as a potent angiogenic factor [50]. Leptin acts both centrally and peripherally to exert the above mentioned effects. Centrally, at hypothalamus, leptin can interact with six types of different receptors that are present on hypothalamic nuclei in order to counteract the effect of neuropeptide Y on mediobasal aspect of arcuate nucleus as potent hunger stimulator and activating alfa-melanocyte-stimulating hormone on lateral aspect of arcuate nucleus as hunger suppressant. In the recent work of Wagner et al, the triggering effect of TLR-4 mediated activation of these receptors and disruption of leptin, ultimately leading to regulation of feeding behavior [51]. In fact a hypothalamic inflammation induced by TLR-4 activation (see below), is possibly involved in leptin resistance and eating behavior as a novel underlying mechanism of obesity [51].
Resistin
Resistin is an adipose-derived peptide hormone encoded by the RETN gene and consists of 180 amino acids, rich in cysteine with molecular weight of 12.5kDa, secreted by the adipose tissue in rodents and by the macrophages in humans.
Resistin inflammatory profile shows that it participates in inflammatory states, such as infection and encounter of bacterial lipopolysaccharide, or chronic conditions like atherosclerosis, lung or liver disease with subsequent increase in expression of pro-inflammatory cytokines such as IL-1, IL-6, IL-12, and TNF alpha. The other inflammatory function of resistin is upregulation of ICAM-1 and VCAM-1 in the recruitment of leukocytes to the site of infection [52,53].
Resistin's correlation with obesity and insulin resistance has been demonstrated in many studies and is the subject of more controversies. It has been demonstrated that serum resistin level is increased in obesity and central obesity is one the major contributing factors [52,54]. Also, it has been shown that leptin administration decreases resistin expression and subsequently glucose homeostasis [55]. Altogether, resistin seems to exert its role in adiposity by inhibiting adipocyte differentiation and ectopic fat accumulation of non-adipose tissue leading to development of type 2 diabetes. But there are robust evidence that overexpression of resistin impairs insulin action in insulin sensitive tissues. The role and contribution of TLR-4 in the pathogenesis of obesity and insulin resistance in association of resistin has been demonstrated [56].
Excess Visceral Adipose Tissue Induces Insulin Resistance
In 1963, Randall proposed that free fatty acids (FFA) inhibits glucose use, and increases production of acetyl co-A, which in turn inhibits mitochondrial function of pyruvate kinase in muscle. In obesity and type 2 diabetes, there is decrease glucose response to insulin. In normal metabolic state, insulin increases uptake of glucose by muscle, adipose tissue and suppresses gluconeogenesis from the liver and blocks glucose output from the liver. In adipose [57] tissue, insulin stimulates LPL, activating clearance of triglycerides rich lipoproteins to FFA and monoglycerides, while suppresses hormone sensitive lipase, the enzymes that is responsive to glucagon, adrenalin and glucocorticoids, and causes release of FFA. However, there is an increased release of FFA in the portal and systemic circulation in the context of obesity and type 2 diabetes, where TNF acts as an insulin antagonist, stimulating the release of FFA which in turns inhibit glucose uptake by their insulin sensitive tissue [33]. In fact, the failure of insulin to suppress FFA release is the early event in the process of insulin resistance. Increase FFA leads to impaired insulin signaling as well. Concomitant to increase TNF secretion impairing insulin function, the decrease in adiponectin level in viscerally obese person also play a crucial role for deepening insulin resistance. As described above, adiponectin suppress fatty acid synthesis, decreases the activity of Acetyl Co-A carboxykinase in liver and muscle, consequently improving lipid oxidation; concomitantly increases free fatty acid beta oxidation through Carnitine palmitoyl transferase, while inhibiting TNF alpha and inflammatory cytokine release and suppressing CD36 as mediator of fatty acid uptake in hepatocytes, preventing both steatohepatosis and steatohepatitis. Inadequate adiponectin and development of leptin resistance further promote both visceral fat accumulation and aggravation of insulin resistance.
Skeletal muscle accounts for 80-90% of insulin mediated glucose disposal [58] and is affected by insulin resistance; however, the more important element in metabolic disturbances is the inability of insulin to block FFA release from adipose tissue and the inability of insulin to block hormone sensitive lipase, which is stimulated by counter-regulatory hormones.
Increase in FFA, which is taken up by myocytes, impairs insulin stimulated glucose disposal in skeletal muscle [59] through impaired phosphatidylinositol-3-kinase (PI3K) phosphorylation, inhibits insulin suppression of glucose production by the liver (glycogenolysis) [60] and causes compensatory increase in Beta cell insulin release to overcome the insulin-mediated glucose disposal defect. In addition, excess FFA promotes hepatic neoglucogenesis [61] and formation of triglycerides in hepatocytes, and release of VLDL from the liver and hepatic glucose output. FFA excess by itself inhibits endothelial LPL [62] as well. In fact, as a result of excess TNF alpha, FFA release has been shown to start well before development of type 2 diabetes [63].
Taken together, the inflammatory process by virtue of inflammatory cytokines that started in adipose tissue promotes FFA release, causes insulin function defect in term of glucose disposal, inability to block glucose output from the liver, and inhibits FFA release from the adipose tissue resulting ultimately in lipo- and gluco-toxicity, which leads to beta cell failure and diabetes. The impaired insulin signaling is the hallmark of insulin resistance.
Molecular Aspect of Impaired Insulin Signaling
In the early-90s, with the discovery of the role of skeletal muscle in insulin resistance some molecules emerged as likely candidate of insulin signaling defect, e.g. insulin receptor itself, insulin related phosphatases, insulin receptor substrates dysfunction, glucokinases, glycogen synthases, and glucose transporters [64]. Both the oxidative glucose utilization and non-oxidative glucose utilization (glycogen synthesis) as well as glucose transport are impaired during early phase of prediabetes [65]. While the binding of insulin to insulin receptor is unchanged, the insulin receptor substrate and insulin receptor kinases in patient with type 2 diabetes does not respond to the same extent as a normal person. Human insulin receptor occurs in two insulin isoforms with type A and B. A normal person has only one isoforms of insulin receptor exclusively, where as diabetic person has both isoforms [66]. The role of regulatory influence from hyperglycemia and TNF factor on insulin receptor isoforms has been shown in skeletal muscle. In fact, both acute hyperglycemia and TNF exert inhibitory effect on insulin signaling, specially on insulin receptor kinase, induced by protein kinase C activation [65].
Insulin binding to insulin receptor alpha subunit induces conformational changes in the beta subunit, which leads to tyrosine phosphorylation of downstream intracellular proteins as shown in (Figure 2) [67]. It was shown that impaired tyrosine phosphorylation was one of the molecular mechanisms of insulin resistance. TNF alpha diminishes tyrosine phosphorylation of insulin receptor substrate 1 (IRS-1) and promotes serine residue phosphorylation. The shift from tyrosine to serine phosphorylation by inflammatory cytokines shifts the metabolic pathways mediated by phosphatidylinositol kinase to mitogenic pathway and MAP kinase pathway [68]. In mouse model of obesity, blocking TNF restores insulin sensitivity, and PPAR Gamma activator reverses TNF mediated IRS-1 tyrosine phosphorylation inhibition [68]. Furthermore, TNF has the ability to inhibit insulin signaling through P55 and P75 TNF receptors by increasing sphingomyelinase activity which converts sphingomylein to ceramides and cholin; an increase in intracellular ceramide, similar to TNF alpha, ultimately suppresses insulin induced tyrosine residue phosphorylation of the insulin receptor and IRS-1 [69]. Thus, providing a molecular mechanism of insulin resistance in inflammatory syndrome associated obesity.
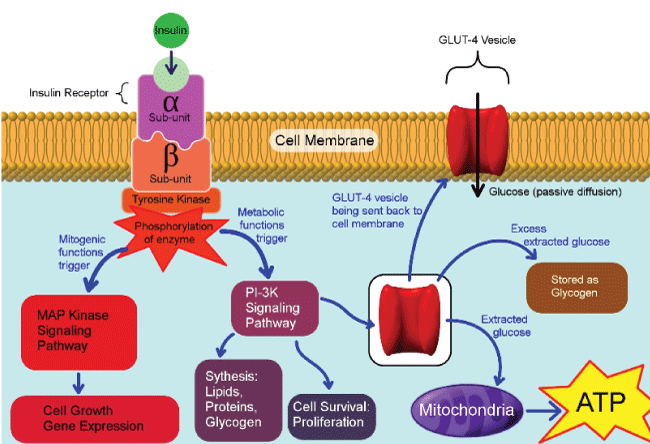
.
Figure 2: Pathways involve after insulin binding to its insulin receptor. The metabolic pathway is mediated through PI3K and mitogenic function mediated by mitogen activated protein (MAP) kinase pathway. The metabolic pathway stimulates glucose uptake through activation of glucose transporter-4 recruitment to cell membrane, along with lipid synthesis in adipose tissue, protein synthesis in most organs and glycogen synthesis in liver and kidneys.
View Figure 2
Part 2 - Why Excess of Natural Fat is Associated with Inflammation?
Innate Immunity
When chemicals from non-living or living prokaryotes are exposed to an organism of higher evolutionary state, a set of reactions follows; the reaction seems to be a product of evolutionary experience of the host. The primordial response to a foreign antigen is called "The Innate Immunity Response", it is mostly reactionary, pattern recognition/signature recognition; however, it evolves to direct the host organism to develop a more sophisticated response (in case the host organism achieved evolutionary steps) to generate the so called adaptive response to target the invasive microorganism in a more specific manner. Though in its primitive form, the innate immunity is still able to distinguish self, commensal bacteria from invasive pathogens and cellular debris from necrosis or apoptosis. The reaction sometimes is costly, highly destructive, and may cause, in certain condition, a massive cytokine storm leading to septic shock.
In humans, the innate immunity is comprised of different modality of responses. Cells and proteins residing in mucosal membrane and skin act as a barrier with ability to distinguish between commensal bacteria versus pathogens. Receptive cells such as phagocytes, dendritic cells, mediate their defensive function by their non specific recognition receptors. The effector cytokines secreted from receptive cells are composed of different degrees of inflammatory activation based on the type and amount of cytokines released with the acute phase protein secretion from the liver and the complement system activation with activation of inflammatory response. Furthermore an array of cells initially known as dendritic cells and natural killer cells engage and promote host defense. Other cells of innate immunity such as basophils, eosinophils and mast cells are to defend cells, or limit the degree of exposure of the living organism to parasites.
The key to the function of innate immunity are TLRs. Various non-communicable diseases such as obesity, and its byproduct, type 2 diabetes, coronary artery diseases, and Alzheimer's Dementia, the so-called "cardiometabolic syndromes" are associated with increased serum cytokines and c-reactive proteins. The interplay of dysmetabolic condition and innate immunity is related to activation of TLR, specifically TLR4, which set the stage for a chain of inflammatory and consequently, an array of metabolic response to support the cost of inflammation, setting another stage for more chaotic cell response, which may emerge down the road as certain cancers.
The Toll Like Receptors
Receptors in innate immunity
Receptor's encounter with an antigen can result in activation of innate immunity or building tolerance and in some cases ignorance of antigen. The initial receptor binding response in innate immunity is based on evolutionary preservation of a memory to recognize certain characteristics of antigen, preserved from ancient time and evolve to react accordingly. It is non-specific, has pattern recognition elements and is able to react to extremely wide array of antigens.
Receptors attached to the cells are categorized as pattern recognition receptors which recognize pathogens associated molecular patterns. The type of receptor that recognizes the antigen and the cell that encounters the antigen are two elements with important ramifications that dictate processes such as secretion of cytokines or opsonins, and invitation of phagocytes and other elements to the scene of encounter.
The major cellular membrane receptor known to be involved in the innate immunity is TLR, which is comprised of 10 different receptors each responsible for pattern recognition with TLR4 as the most notable receptor. A list summarizing TLR's physiological functions, their target ligand, the most common target e.g. microbial or endogenous pathological condition associated with their activation, and their co-receptors site are listed in table 1. TLR's residence on different immune cells and the known pathological response or pathological condition associated with their activation is summarized in table 2. Apart from TLRs, there is a set of receptors known as scavenger receptors, macrophage mannose receptors and Dectin-1 as well.
![]()
Table 1: Major Toll like Receptors characteristics, functions and targets
View Table 1
![]()
Table 2: Toll-Like co-receptor and their characterization
View Table 2
The innate immunity can execute its function in the following ways: 1) express on the surface of cells such as Toll Like transmembrane receptors; 2) free flowing soluble receptors such as c-lectin molecules either bound to membrane or soluble to recognize certain glycoprotein associated to pathogenic antigens; or 3) being cytosolic receptors, where they can recognize DNAs or phagocytosed antigens. The innate immunity can also be categorized based on their function and types of antigens recognized in the following categories: 1) foreign pathogenic antigen (antimicrobial response), 2) foreign non-pathogenic antigens (allergens, obesity), 3) self-antigen (autoimmunity), 4) altered self-antigen (cancers), and 5) foreign non-pathogen tolerance (commensal bacteria) [70].
Co-receptor of innate immunity and signaling pathways
Innate immunity, mostly through TLR, direct the first line of defense against invading antigens, intracellular DNAs, and large number of host and exogenous antigens by pattern recognitions, setting the stage for more specific approach against the pathogen or perceived pathogen (e.g. anti-DNA in Systemic Lupus Erythematosus). This highly conserved pathogen-associated recognition molecules necessitates co-receptor for activation leading to cytokines or chemokines secretion through signal transduction, or in the opposite end, to prevent activation when exposed to self-antigens or antigens that needs to stay and promote biological entity proliferation (sperm) without causing harm to the antigen or surrounding tissues. Table 2 summarizes the co-receptor associated with each TLR.
The function of TLR4 in sensing lipopolysaccharide (LPS) of bacteria, which is a prototypical sensing machinery of TLRs. The acute phase protein, Lipoprotein Binding Protein (LBP), binds to LPS and transfer the LPS to the membrane anchored TLR by mediation of CD14. This crucial part of transfer of LPS is a product of different level of interaction that starts from the concentration of LBP, the ligand co-receptor interaction which triggers PKC to activate PI (4,5) p2 to start a chain of phosphorylation at cellular membrane, endosomal membrane, or simply a lipid raft, to trigger the formation NF kappa B and AP1. The seminal discoveries of TLR4 and its associated co-receptor MD-2 contributed to the establishment of a model in which CD14 acts as an LPS sensing receptor that accepts LPS monomers from LBP and then transfer to TLR4:MD2 complex, thereby promoting its ligand-induced dimerization.
Acute phase reactant, LBP in high concentration inhibits LPS recognition, protecting the host from LPS-or gram-negative septic shock. Interestingly, it seems that LBP exert this inhibitory action at least partly by transferring LPS to serum Lipoproteins, such as HDL or by forming inactive aggregates [71].
LBP acts similarly in the activation of TLR once exposed to the product of adipose tissue lipids (e.g. palmytoil) in chronic obesity. Decreased level of HDL in visceral obesity and during the process of diabetogenesis is thought to be due to rapid clearance of HDL, but factor as described above may explain the rapid clearance of the lipoproteins from circulation, to prevent further TL4 activation, and acts as compensatory mechanism.
Toll-like receptor 4: The link between obesity and inflammation
As discussed earlier, viscerally obese subjects have activated pro-inflammatory signals, increased downstream markers of inflammation, activated anti-inflammatory signals to counteract the inflammation, and hyperglycemia as a byproduct due to insulin resistance induced by cytokines and beta cell failure. The question is why do we have an inflammatory syndrome when we have an excess of visceral adiposity and excess of circulating FFA? And what is the link?.
TLR-4 is known to bind to LPS of gram-negative bacterial cell walls [72,73]. Upon binding to LPS, its co-receptors trigger a downstream signaling cascade leading to activation of NF Kappa B pathway, which then activates the transcription of many proinflammatory genes that encodes proinflammatory meolecules, including cytokines, chemokines and other effectors of innate immune response [73]. The majority of LPS is acylated with a moiety (Lipid A) that is saturated fatty acids and removal of the Lipid A results in complete loss of endotoxic activity.
Further research revealed that TLR4 is able to profile palmitic, stearic and lauric acid of FFA [74,75]. LPA and lauric acid enhances the association of TLR-4 with MD2 and downstream adaptor molecules, MyD and TRIF, lead to activation of NF-kappa B [76]. Interestingly, hexadecanoic acid and other polyunsaturated fatty acids downregulate TLR-4 activation. Palmitate and oleate, two of the most abundant nutritional fatty acids, can also activate TLR-4 signaling [77]. Activation of Nf Kappa B activates the transcription of many proinflammatory genes [78]. It has been shown that FFA activates NF Kappa B in macrophage cell lines and causes cytokine expression via TLR4 signaling. Not only lauric acid, but medium chain fatty acids, palmitate and oleate, can also activate TLR-4 of macrophages. Similarly, FFA activates TLR4 signaling in adipose cells and tissue from cell culture to animal model of ob/ob, db/db and diet induced obese mice, demonstrating adipose tissue is functional and responding to excess adipose tissue [77]. The TLR-4 knockdown mice adipocytes, once treated with FFA of palmitate and oleates, cannot generate TNF alpha and IL-6, confirming the central role of TLR on FFA induced inflammatory response.
As described earlier, FFA have been shown to cause insulin resistance in vivo, and it appears that fatty acid induced insulin resistance may be mediated in part by the proinflammatory signals. How the FFA are sensed by adipose tissue in order to activate inflammatory signal is unknown.
It is thought in humans that acute elevation of FFA activates inflammatory signals through adipocytes mediated by TLR-4 signaling pathway [79]. Recently, a monoclonal antibody against fatty acid lipid binding protein was described [80]. Fatty acid lipid binding proteins are a homologous polypeptide of 14-15KD protein that facilitates bindings of lipids to lipid membrane vesicle. It facilitates solubilization and lipid transfer in adipocytes and has been found as essential factor in adipose tissue metabolic response [81]. It is the most abundant protein in adipose tissue and was considered a possible target in adipose inflammatory disease e.g. obesity-diabetes. The monoclonal antibody against fatty acid lipid binding protein has been shown to attenuate the inflammatory response (TNF alpha, IL-6 and MCP-1) through inhibition of TLR4 in diet-induced obese mouse model [80] and could play a crucial role in the development of treatment for obesity/inflammation induced type 2 diabetes.
High fat diet in both wild animal mice and TLR null mice causes weight gain and similar fat accumulation. Wild animal elicits inflammatory cytokine response in the aortic lysate with activation of IKK Beta upstream of the signaling cascade, whereas TLR-4 null (TLR-4 -/-) mice does not demonstrate inflammatory cytokine expression, without activation Ikk Beta activity, and no down stream activation of universal marker of cellular inflammation: the NF Kappa B activation. In wild animal mice, insulin induced phosphorylation of eNOS (a unique aspect of endothelium) takes place, as compared to TLR 4 null mice which has aborted response [82]. This landmark study clearly demonstrated that NFkappa B is a mediator of activation of TLR-4 receptor in endothelial tissue. In human microvascular endothelial cells, free fatty acid palmitate, subsequent to the activation of TLR-4 mediated IKK beta and downstream activation of NF Kappa B, causes IL-6 and ICAM mediated insulin resistance. Insulin resistance can be blocked by inhibiting TLR-4 function by siRNA or through My 88 (TLR-4 co-receptor) blockage [82]. These results demonstrated NF Kappa B is downstream of TLR-4 receptor signaling pathway [82].
Role of Glucocorticoids in Obesity and Inflammation
Since the description of obesity as a "Cushing's syndrome of omentum" [83] by Stewart and colleagues, much is known now that elicit the detrimental anti-inflammatory response to obesity. As readers may remember, 11 beta hydroxysteroid dehydrogenase type 1 (11BHSD1) is crucial for converting hydrocortisone which is inactive glucocorticoid to active cortisol. There is abundance of type 1 oxydoreductase enzyme in omentum and visceral adipose tissue. The consequence of excess visceral adipose tissue is the localized production of active glucocorticoid (cortisol) in the adipose tissue, which generates local anti-inflammatory effect; however, the most detrimental effect is due to release of FFA from visceral adipose tissue depots, contributing to excess fatty acid release to the portal circulation and ultimately, insulin resistance and abnormal glucose levels [83]. In fact, the 11BHSD1mRNA expression of omental adipose tissue is 13 times higher in obese subjects when compared to normal healthy person [84]. However there is no difference in visceral adipose tissue 11BHSD1 mRNA enzyme of control subjects and patients with Cushing's syndrome due cortisol secreting adrenal adenomas [84]. It is known that in obese Zucker rats, 11BHSD1 is increased in adipose tissue and decreased in liver [85] while there is an increased production of corticosterone and 11 oxidated substrate. Similarly, obese person have excess 24 hour daily cortisol production and excretion compared to normal weight person [86]; however, the serum free cortisol level is not different owing to the increased cortisol metabolization rate. Men have higher cortisol production rate compared to women, and the excess production is thought to be due to excess visceral fat in men compared to women [87] and increases with age. There is a strong relation between intrabdominal fat as measured by CAT scan and cortisol production rate measured by dilution isotopes [86]. What is surprising is with weight loss, the cortisol production rate as measured in crude term or adjusted for fat mass or intrabdominal fat is increased by 40% and 100% [86], and the increase in production is during the day as oppose to patient with Cushing's disease, where as the 11BHSD1 enzyme expression decreases with weight loss. Insulin sensitivity correlates with increase cortisol production and whether this is direct or indirect effect of excess adipose fat, the increase activity of 11BHSD1 may play a localize mechanism against inflammation in the visceral adipose tissue, which by itself causes further accumulation of visceral fat and promote insulin resistance and decline of insulin secretion leading to beta cell failure. The increase in 11BHSD activity with weight gain seems to be a consequence of the visceral adiposity. The alteration and activation of adrenocorticotopic axis in obesity contribute to the progression of obesity and diabetes.
The antihyperglycemic effect of metformin is due to decrease neoglucogenesis in the liver, though the well known effect of metformin on phosphorylation of AMP activated protein kinase relationship to glucose metabolism is unclear. However, in a recent study on metabolomic of metformin in healthy subjects demonstrated an involvement of neuroendocrine system by change in urine cortisol. In fact, metformin was found to induce AMPK liver X receptor alpha phosphorylation followed interestingly by proopiomelancortin suppression in rat pituitary cells. The decline in 24 hour urine cortisol after metformin has been reported in humans and explains why metformin decreases neoglucogenesis, the effect that is associated with suppression of pituitary adrenal axis [88].
Conclusion
Visceral obesity is a precursor of an activation of inflammatory process, the link of obesity to the inflammatory system is activation of TLR-4 by FFA release induced by the cytokines due to the activation of adipocytes, and macrophages residing in adipose tissue. The contribution of 11BHSD as an anti-inflammatory factor in visceral obesity contribute further to FFA release together leading to insulin resistance and visceral fat gain, which ultimately leads to development of type 2 diabetes. The inflammatory metabolic syndrome has to be prevented in early phase of obesity to avoid diabetes and its complication.
Acknowledgement
We would like to extend our appreciation to Dr. Henri Lin for scientific editing of the manuscript.
Conflicts of Interest
Received Consultation fee from Eisai. Inc.
References
-
Small M, Lowe GD, MacCuish AC, Forbes CD (1987) Thrombin and plasmin activity in diabetes mellitus and their association with glycaemic control. Q J Med 65: 1025-1031.
-
Davi G, Violi F, Catalano I, Giammarresi C, Putignano E, et al. (1991) Increased plasminogen activator inhibitor antigen levels in diabetic patients with stable angina. Blood Coagul Fibrinolysis 2: 41-45.
-
Schneider SH, Kim HC, Khachadurian AK, Ruderman NB (1988) Impaired fibrinolytic response to exercise in type II diabetes: effects of exercise and physical training. Metabolism 37: 924-929.
-
Ford I, Singh TP, Kitchen S, Makris M, Ward JD, et al. (1991) Activation of coagulation in diabetes mellitus in relation to the presence of vascular complications. Diabet Med 8: 322-329.
-
Aso Y, Matsumoto S, Fujiwara Y, Tayama K, Inukai T, et al. (2002) Impaired fibrinolytic compensation for hypercoagulability in obese patients with type 2 diabetes: association with increased plasminogen activator inhibitor-1. Metabolism 51: 471-476.
-
Lim HS, Chong AY, Freestone B, Blann AD, Lip GY (2005) The effect of multi-factorial intervention on plasma von Willebrand factor, soluble E-selectin and tissue factor in diabetes mellitus: implications for atherosclerotic vascular disease. Diabet Med 22: 249-255.
-
Bagg W, Ferri C, Desideri G, Gamble G, Ockelford P, et al. (2001) The influences of obesity and glycemic control on endothelial activation in patients with type 2 diabetes. J Clin Endocrinol Metab 86: 5491-5497.
-
Aoki I, Shimoyama K, Aoki N, Homori M, Yanagisawa A, et al. (1996) Platelet-dependent thrombin generation in patients with diabetes mellitus: effects of glycemic control on coagulability in diabetes. J Am Coll Cardiol 27: 560-566.
-
Osende JI, Badimon JJ, Fuster V, Herson P, Rabito P, et al. (2001) Blood thrombogenicity in type 2 diabetes mellitus patients is associated with glycemic control. J Am Coll Cardiol 38: 1307-1312.
-
Tsai JS, Chuang LM, Chen CS, Liang CJ, Chen YL, et al. (2014) Troglitazone and Delta2Troglitazone enhance adiponectin expression in monocytes/macrophages through the AMP-activated protein kinase pathway. Mediators Inflamm 2014: 726068.
-
Jiang C, Ting AT, Seed B (1998) PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature 391: 82-86.
-
Wellen KE, Hotamisligil GS (2003) Obesity-induced inflammatory changes in adipose tissue. J Clin Invest 112: 1785-1788.
-
Ziccardi P, Nappo F, Giugliano G, Esposito K, Marfella R, et al. (2002) Reduction of inflammatory cytokine concentrations and improvement of endothelial functions in obese women after weight loss over one year. Circulation 105: 804-809.
-
Beutler B, Mahoney J, Le Trang N, Pekala P, Cerami A (1985) Purification of cachectin, a lipoprotein lipase-suppressing hormone secreted by endotoxin-induced RAW 264.7 cells. J Exp Med 161: 984-995.
-
MacPherson GG and North RJ (1986) Endotoxin-mediated necrosis and regression of established tumours in the mouse. A correlative study of quantitative changes in blood flow and ultrastructural morphology. Cancer Immunol Immunother 21: 209-216.
-
Nawroth PP, Stern DM (1986) Modulation of endothelial cell hemostatic properties by tumor necrosis factor. J Exp Med 163: 740-745.
-
Schleef RR, Bevilacqua MP, Sawdey M, Gimbrone MA Jr, Loskutoff DJ (1988) Cytokine activation of vascular endothelium. Effects on tissue-type plasminogen activator and type 1 plasminogen activator inhibitor. J Biol Chem 263: 5797-5803.
-
Simpson SQ, Casey LC (1989) Role of tumor necrosis factor in sepsis and acute lung injury. Crit Care Clin 5: 27-47.
-
Zwaginga JJ, Sixma JJ, and de Groot PG (1990) Activation of endothelial cells induces platelet thrombus formation on their matrix. Studies of new in vitro thrombosis model with low molecular weight heparin as anticoagulant. Arteriosclerosis 10: 49-61.
-
Clauss M, Gerlach M, Gerlach H, Brett J, Wang F, et al. (1990) Vascular permeability factor: a tumor-derived polypeptide that induces endothelial cell and monocyte procoagulant activity, and promotes monocyte migration. J Exp Med 172: 1535-1545.
-
Paleolog EM, Delasalle SA, Buurman WA, Feldmann M (1994) Functional activities of receptors for tumor necrosis factor-alpha on human vascular endothelial cells. Blood 84: 2578-2590.
-
22. Stern DM, Esposito C, Gerlach H, Gerlach M, Ryan J, et al. (1991) Endothelium and regulation of coagulation. Diabetes Care 14: 160-166.
-
Yudkin JS, Panahloo A, Stehouwer C, Emeis JJ, Bulmer K, et al. (2000) The influence of improved glycaemic control with insulin and sulphonylureas on acute phase and endothelial markers in Type II diabetic subjects. Diabetologia 43: 1099-1106.
-
Yudkin JS (2003) Adipose tissue, insulin action and vascular disease: inflammatory signals. Int J Obes Relat Metab Disord 27 Suppl 3: S25-28.
-
Yudkin JS (1999) Abnormalities of coagulation and fibrinolysis in insulin resistance. Evidence for a common antecedent? Diabetes Care 22 Suppl 3: C25-30.
-
Semb H, Peterson J, Tavernier J, Olivecrona T (1987) Multiple effects of tumor necrosis factor on lipoprotein lipase in vivo. J Biol Chem 262: 8390-8394.
-
Kern PA (1988) Recombinant human tumor necrosis factor does not inhibit lipoprotein lipase in primary cultures of isolated human adipocytes. J Lipid Res 29: 909-914.
-
Feingold KR, Grunfeld C (1987) Tumor necrosis factor-alpha stimulates hepatic lipogenesis in the rat in vivo. J Clin Invest 80: 184-190.
-
Hotamisligil GS, Shargill NS, Spiegelman BM (1993) Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 259: 87-91.
-
Sartipy P, Loskutoff DJ (2003) Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proc Natl Acad Sci U S A 100: 7265-7270.
-
Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, et al. (2003) Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 112: 1796-1808.
-
Xu H, Barnes GT, Yang Q, Tan G, Yang D, et al. (2003) Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 112: 1821-1830.
-
Christiansen T, Richelsen B and Bruun JM (2005) Monocyte chemoattractant protein-1 is produced in isolated adipocytes, associated with adiposity and reduced after weight loss in morbid obese subjects. Int J Obes (Lond) 29: 146-50.
-
Xing Z, Gauldie J, Cox G, Baumann H, Jordana M, et al. (1998) IL-6 is an antiinflammatory cytokine required for controlling local or systemic acute inflammatory responses. J Clin Invest 101: 311-320.
-
de Mello VD, Kolehmainen M, Schwab U, Mager U, Laaksonen DE, et al. (2008) Effect of weight loss on cytokine messenger RNA expression in peripheral blood mononuclear cells of obese subjects with the metabolic syndrome. Metabolism 57: 192-199.
-
Ghanim H, Aljada A, Hofmeyer D, Syed T, Mohanty P, et al. (2004) Circulating mononuclear cells in the obese are in a proinflammatory state. Circulation 110: 1564-1571.
-
Carey AL, Steinberg GR, Macaulay SL, Thomas WG, Holmes AG, et al. (2006) Interleukin-6 increases insulin-stimulated glucose disposal in humans and glucose uptake and fatty acid oxidation in vitro via AMP-activated protein kinase. Diabetes 55: 2688-2697.
-
Yoon MJ, Lee GY, Chung JJ, Ahn YH, Hong SH, et al. (2006) Adiponectin increases fatty acid oxidation in skeletal muscle cells by sequential activation of AMP-activated protein kinase, p38 mitogen-activated protein kinase, and peroxisome proliferator-activated receptor alpha. Diabetes 55: 2562-2570.
-
Dyck DJ, Heigenhauser GJ, Bruce CR (2006) The role of adipokines as regulators of skeletal muscle fatty acid metabolism and insulin sensitivity. Acta Physiol (Oxf) 186: 5-16.
-
Gil-Campos M, Canete RR, Gil A (2004) Adiponectin, the missing link in insulin resistance and obesity. Clin Nutr 23: 963-974.
-
Kissebah AH, Sonnenberg GE, Myklebust J, Goldstein M, Broman K, et al. (2000) Quantitative trait loci on chromosomes 3 and 17 influence phenotypes of the metabolic syndrome. Proc Natl Acad Sci U S A 97: 14478-14483.
-
Kadowaki T, Yamauchi T (2005) Adiponectin and adiponectin receptors. Endocr Rev 26: 439-451.
-
Vozarova B, Stefan N, Lindsay RS, Krakoff J, Knowler WC, et al. (2002) Low plasma adiponectin concentrations do not predict weight gain in humans. Diabetes 51: 2964-2967.
-
Tsuchida A, Yamauchi T, Ito Y, Hada Y, Maki T, et al. (2004) Insulin/Foxo1 pathway regulates expression levels of adiponectin receptors and adiponectin sensitivity. J Biol Chem 279: 30817-30822.
-
Yamaguchi N, Argueta JG, Masuhiro Y, Kagishita M, Nonaka K, et al. (2005) Adiponectin inhibits Toll-like receptor family-induced signaling. FEBS Lett 579: 6821-6826.
-
Jenke A, Wilk S, Poller W, Eriksson U, Valaperti A, et al. (2013) Adiponectin protects against Toll-like receptor 4-mediated cardiac inflammation and injury. Cardiovasc Res 99: 422-431.
-
You M, Rogers CQ (2009) Adiponectin: a key adipokine in alcoholic fatty liver. Exp Biol Med (Maywood) 234: 850-859.
-
Conde J, Scotece M, Gómez R, López V, Gómez-Reino JJ, et al. (2011) Adipokines: biofactors from white adipose tissue. A complex hub among inflammation, metabolism, and immunity. Biofactors 37: 413-420.
-
Green ED, Maffei M, Braden VV, Proenca R, DeSilva U, et al. (1995) The human obese (OB) gene: RNA expression pattern and mapping on the physical, cytogenetic, and genetic maps of chromosome 7. Genome Res 5: 5-12.
-
Tahergorabi Z, Khazaei M (2015) Leptin and its cardiovascular effects: Focus on angiogenesis. Adv Biomed Res 4: 79.
-
Reis WL, Yi CX, Gao Y, Tschöp MH, Stern JE (2015) Brain innate immunity regulates hypothalamic arcuate neuronal activity and feeding behavior. Endocrinology 156: 1303-1315.
-
Olefsky JM, Glass CK (2010) Macrophages, inflammation, and insulin resistance. Annu Rev Physiol 72: 219-246.
-
Mojiminiyi OA, Abdella NA (2007) Associations of resistin with inflammation and insulin resistance in patients with type 2 diabetes mellitus. Scand J Clin Lab Invest 67: 215-225.
-
Verma S, Li SH, Wang CH, Fedak PW, Li RK, et al. (2003) Resistin promotes endothelial cell activation: further evidence of adipokine-endothelial interaction. Circulation 108: 736-740.
-
Asensio C, Cettour-Rose P, Theander-Carrillo C, Rohner-Jeanrenaud F, Muzzin P (2004) Changes in glycemia by leptin administration or high- fat feeding in rodent models of obesity/type 2 diabetes suggest a link between resistin expression and control of glucose homeostasis. Endocrinology 145: 2206-13.
-
Tarkowski A, Bjersing J, Shestakov A, Bokarewa MI (2010) Resistin competes with lipopolysaccharide for binding to toll-like receptor 4. J Cell Mol Med 14: 1419-1431.
-
Ruan H, Lodish HF (2003) Insulin resistance in adipose tissue: direct and indirect effects of tumor necrosis factor-alpha. Cytokine Growth Factor Rev 14: 447-455.
-
DeFronzo RA, Gunnarsson R, Björkman O, Olsson M, Wahren J (1985) Effects of insulin on peripheral and splanchnic glucose metabolism in noninsulin-dependent (type II) diabetes mellitus. J Clin Invest 76: 149-155.
-
Kruszynska YT, Worrall DS, Ofrecio J, Frias JP, Macaraeg G, et al. (2002) Fatty acid-induced insulin resistance: decreased muscle PI3K activation but unchanged Akt phosphorylation. J Clin Endocrinol Metab 87: 226-234.
-
Boden G, Cheung P, Stein TP, Kresge K, Mozzoli M (2002) FFA cause hepatic insulin resistance by inhibiting insulin suppression of glycogenolysis. Am J Physiol Endocrinol Metab 283: E12-19.
-
Staehr P, Hother-Nielsen O, Landau BR, Chandramouli V, Holst JJ, et al. (2003) Effects of free fatty acids per se on glucose production, gluconeogenesis, and glycogenolysis. Diabetes 52: 260-267.
-
Saxena U, Witte LD, Goldberg IJ (1989) Release of endothelial cell lipoprotein lipase by plasma lipoproteins and free fatty acids. J Biol Chem 264: 4349-4355.
-
Ruan H, Miles PD, Ladd CM, Ross K, Golub TR, et al. (2002) Profiling gene transcription in vivo reveals adipose tissue as an immediate target of tumor necrosis factor-alpha: implications for insulin resistance. Diabetes 51: 3176-3188.
-
Haring, HU and Mehnert H (1993) Pathogenesis of type 2 (non-insulin-dependent) diabetes mellitus: candidates for a signal transmitter defect causing insulin resistance of the skeletal muscle. Diabetologia 36: 176-182.
-
Kellerer M, Häring HU (1995) Pathogenesis of insulin resistance: modulation of the insulin signal at receptor level. Diabetes Res Clin Pract 28 Suppl: S173-177.
-
Mosthaf L, Vogt B, Häring HU, Ullrich A (1991) Altered expression of insulin receptor types A and B in the skeletal muscle of non-insulin-dependent diabetes mellitus patients. Proc Natl Acad Sci U S A 88: 4728-4730.
-
Häring HU (1991) The insulin receptor: signalling mechanism and contribution to the pathogenesis of insulin resistance. Diabetologia 34: 848-861.
-
Kanety H, Feinstein R, Papa MZ, Hemi R, Karasik A (1995) Tumor necrosis factor alpha-induced phosphorylation of insulin receptor substrate-1 (IRS-1). Possible mechanism for suppression of insulin-stimulated tyrosine phosphorylation of IRS-1. J Biol Chem 270: 23780-23784.
-
Peraldi P, Hotamisligil GS, Buurman WA, White MF, Spiegelman BM (1996) Tumor necrosis factor (TNF)-alpha inhibits insulin signaling through stimulation of the p55 TNF receptor and activation of sphingomyelinase. J Biol Chem 271: 13018-13022.
-
Liu D, Rhebergen AM, Eisenbarth SC (2013) Licensing Adaptive Immunity by NOD-Like Receptors. Front Immunol 4: 486.
-
Ostuni R, Zanoni I, Granucci F (2010) Deciphering the complexity of Toll-like receptor signaling. Cell Mol Life Sci 67: 4109-4134.
-
Janeway CA Jr, Medzhitov R (2002) Innate immune recognition. Annu Rev Immunol 20: 197-216.
-
Medzhitov R (2001) Toll-like receptors and innate immunity. Nat Rev Immunol 1: 135-145.
-
Hwang D (2001) Modulation of the expression of cyclooxygenase-2 by fatty acids mediated through toll-like receptor 4-derived signaling pathways. FASEB J 15: 2556-2564.
-
Lee JY, Plakidas A, Lee WH, Heikkinen A, Chanmugam P, et al. (2003) Differential modulation of Toll-like receptors by fatty acids: preferential inhibition by n-3 polyunsaturated fatty acids. J Lipid Res 44: 479-486.
-
Wong SW, Kwon MJ, Choi AM, Kim HP, Nakahira K, et al. (2009) Fatty acids modulate Toll-like receptor 4 activation through regulation of receptor dimerization and recruitment into lipid rafts in a reactive oxygen species-dependent manner. J Biol Chem 284: 27384-27392.
-
Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, et al. (2006) TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest 116: 3015-3025.
-
Zuany-Amorim C, Hastewell J, Walker C (2002) Toll-like receptors as potential therapeutic targets for multiple diseases. Nat Rev Drug Discov 1: 797-807.
-
Boden G (2008) Obesity and free fatty acids. Endocrinol Metab Clin North Am 37: 635-646, viii-ix.
-
Miao X, Wang Y, Wang W, Lv X, Wang M, et al. (2015) The mAb against adipocyte fatty acid-binding protein 2E4 attenuates the inflammation in the mouse model of high-fat diet-induced obesity via toll-like receptor 4 pathway. Mol Cell Endocrinol 403: 1-9.
-
Furuhashi M, Fucho R, Görgün CZ, Tuncman G, Cao H, et al. (2008) Adipocyte/macrophage fatty acid-binding proteins contribute to metabolic deterioration through actions in both macrophages and adipocytes in mice. J Clin Invest 118: 2640-2650.
-
Kim F, Pham M, Luttrell I, Bannerman DD, Tupper J, et al. (2007) Toll-like receptor-4 mediates vascular inflammation and insulin resistance in diet-induced obesity. Circ Res 100: 1589-1596.
-
Bujalska IJ, Kumar S, Stewart PM (1997) Does central obesity reflect "Cushing's disease of the omentum"? Lancet 349: 1210-1213.
-
Mariniello B, Ronconi V, Rilli S, Bernante P, Boscaro M, et al. (2006) Adipose tissue 11beta-hydroxysteroid dehydrogenase type 1 expression in obesity and Cushing's syndrome. Eur J Endocrinol 155: 435-441.
-
Livingstone DE, Jones GC, Smith K, Jamieson PM, Andrew R, et al. (2000) Understanding the role of glucocorticoids in obesity: tissue-specific alterations of corticosterone metabolism in obese Zucker rats. Endocrinology 141: 560-563.
-
Purnell JQ, Kahn SE, Samuels MH, Brandon D, Loriaux DL, et al. (2009) Enhanced cortisol production rates, free cortisol, and 11beta-HSD-1 expression correlate with visceral fat and insulin resistance in men: effect of weight loss. Am J Physiol Endocrinol Metab 296: E351-357.
-
Purnell JQ, Brandon DD, Isabelle LM, Loriaux DL, Samuels MH (2004) Association of 24-hour cortisol production rates, cortisol-binding globulin, and plasma-free cortisol levels with body composition, leptin levels, and aging in adult men and women. J Clin Endocrinol Metab 89: 281-287.
-
Cho K, Chung JY, Cho SK, Shin HW, Jang IJ, et al. (2015) Antihyperglycemic mechanism of metformin occurs via the AMPK/LXRα/POMC pathway. Sci Rep 5: 8145.
-
Wetzler LM (2003) The role of Toll-like receptor 2 in microbial disease and immunity. Vaccine 21 Suppl 2: S55-60.
-
Schumann RR, Tapping RI (2007) Genomic variants of TLR1--it takes (TLR-)two to tango. Eur J Immunol 37: 2059-2062.
-
Kirschning CJ, Schumann RR (2002) TLR2: cellular sensor for microbial and endogenous molecular patterns. Curr Top Microbiol Immunol 270: 121-144.
-
Tatematsu M, Seya T, Matsumoto M (2014) Beyond dsRNA: Toll-like receptor 3 signalling in RNA-induced immune responses. Biochem J 458: 195-201.
-
Kyriakidis NC, Kapsogeorgou EK, Tzioufas AG2 (2014) A comprehensive review of autoantibodies in primary Sjögren's syndrome: clinical phenotypes and regulatory mechanisms. J Autoimmun 51: 67-74.
-
Beutler B (2000) Tlr4: central component of the sole mammalian LPS sensor. Curr Opin Immunol 12: 20-26.
-
Eguchi K, Manabe I (2014) Toll-like receptor, lipotoxicity and chronic inflammation: the pathological link between obesity and cardiometabolic disease. J Atheroscler Thromb 21: 629-639.
-
Vijay-Kumar M, Gewirtz AT (2009) Flagellin: key target of mucosal innate immunity. Mucosal Immunol 2: 197-205.
-
Oliveira-Nascimento L, Massari P, Wetzler LM (2012) The Role of TLR2 in Infection and Immunity. Front Immunol 3: 79.
-
Ewald SE, Barton GM (2011) Nucleic acid sensing Toll-like receptors in autoimmunity. Curr Opin Immunol 23: 3-9.
