

International Journal of Diabetes and Clinical Research
Early Release of HMGB1 may Aggravate Neuronal Damage after Transient Focal Ischemia in Diabetic Rat Brain
Naohiro Iwata1, Mari Okazaki2, Shinya Kamiuchi1, Meiyan Xuan3, Hirokazu Matsuzaki2, Takeshi Sakamoto3 and Yasuhide Hibino1*
1Laboratory of Immunobiochemistry, Department of Clinical Dietetics & Human Nutrition, Faculty of Pharmaceutical Sciences, Josai University, Japan
2Laboratory of Pharmacology, School of Pharmaceutical Sciences, Faculty of Pharmaceutical Sciences, Josai University, Japan
3Laboratory of Organic and Medicinal Chemistry, School of Pharmaceutical Sciences, Faculty of Pharmaceutical Sciences, Josai University, Japan
*Corresponding author:
Yasuhide Hibino, Laboratory of Immunobiochemistry, Department of Clinical Dietetics & Human Nutrition, Faculty of Pharmaceutical Sciences, Josai University, 1-1 Keyakidai Sakado Saitama 350-0295, Japan, Tel: +81-49-271-7285, Fax: +81-49-271-7284, E-mail: seitaib@josai.ac.jp
Int J Diabetes Clin Res, IJDCR-2-021, (Volume 2, Issue 1), Research Article; ISSN: 2377-3634
Received: December 03, 2014 | Accepted: February 18, 2015 | Published: February 22, 2015
Citation: Iwata N, Okazaki M, Kamiuchi S, Xuan M, Matsuzaki H, et al. (2015) Early Release of HMGB1 may Aggravate Neuronal Damage after Transient Focal Ischemia in Diabetic Rat Brain. Int J Diabetes Clin Res 2:021. 10.23937/2377-3634/1410021
Copyright: © 2015 Iwata N, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Objective: High Mobility Group Box 1 (HMGB1) released extracellularly from necrotic cells evokes delayed inflammatory processes via interaction with the Receptor for Advanced Glycation End Products (RAGE) or Toll-like Receptors (TLR) in postischemic brain. The diabetic state (DM) aggravated cerebral ischemic injury following the stroke in rats. Therefore, we examined the behavior of HMGB1 and the expression of RAGE in non-DM and DM rat brain after middle cerebral artery occlusion followed by reperfusion (MCAO/Re).
Methods: Diabetes was induced by a single injection of streptozotocin in male Sprague Dawley rats (DM group). MCAO/Re was performed in non-DM and DM rats (ischemic groups) using a standard intraluminal procedure, and postischemic neurological deficits. Both brain infarction and edema were evaluated at various times after reperfusion. Control non-DM and DM rats underwent sham operation using the same manipulation, but without insertion of the occlusion filament. The behavior of HMGB1 and the expression of its receptors in the rat brain were examined using immunohistochemical and western blot analyses.
Results: In sham-operated DM rat brain, immunoreactivity of HMGB1, which was localized in the neuronal nuclei of the cortex, was markedly increased compared with that in non-DM sham-operated rat brain. In the ischemic groups, the DM state aggravated MCAO/Re-induced neurological deficits and cerebral injury assessed by the infarction volume. Enhancement of translocation of HMGB1 from the nucleus to the cytoplasm induced by MCAO/Re was markedly accelerated in the penumbral region of DM rat cortex. Immunoblot analysis revealed that the ischemia-induced increase in the release of HMGB1 into the cerebrospinal fluid and plasma was also enhanced in DM rats. Moreover, the expression of RAGE was upregulated in the brains of DM ischemic and control rats.
Conclusions: The early release of HMGB1 and the expression of its receptors may be involved in the aggravation of neuronal damage caused by transient cerebral ischemia in DM rats. Therefore, it is important to inhibit the HMGB1 released in response to ischemia during the treatment of postischemic injury in diabetic patients.
Keywords
High-mobility group box 1 (HMGB1), Diabetes, Middle cerebral artery occlusion (MCAO), Brain injury, Receptor for advanced glycation end products (RAGE)
Introduction
Diabetes mellitus (DM), which causes chronic hyperglycemia and increases physiological oxidative stress, is a major risk factor for atherosclerotic diseases such as acute brain ischemia [1,2]. World Health Organization data show that about 386 million people worldwide are currently suffering from diabetes. Diabetic patients have a higher risk of stroke compared with nondiabetic patients, and they are more likely to have a poor prognosis and increased mortality after stroke [3,4]. Therefore, it is medically and socially important to reduce the complications of both diabetes and stroke. Previous studies have demonstrated that diabetes also increases oxidative stress in the brain and aggravates cerebral ischemic injury in the animal models [5-7]. In particular, hyperglycemia in the diabetic state increases the formation of reactive oxygen species (ROS) after reperfusion of the blood flow [8,9]. Furthermore, enhanced oxidative stress in the brain induced by diabetic hyperglycemia contributes to the exacerbation of the brain injury caused by transient ischemia [10,11].
High-mobility group box 1 (HMGB1) was first identified as a nonhistone chromosomal protein involved in DNA binding [12]. A few years later, it was isolated as a heparin-binding protein that promotes neurite outgrowth in rat brain and was called amphoterin [13,14]. In 1999, HMGB1 was recognized as a proinflammatory cytokine that mediates endotoxin lethality in mice [15]. HMGB1 is highly conserved through evolution, and has 99% identity among all the mammals. Out of its 215 amino acids, only two residues are substituted in rodent and human versions [16]. HMGB1, a ubiquitous protein present in the nuclei of nearly all the cell types, can be actively secreted by different cell types, including activated monocytes and macrophages [15,17], mature dendritic cells [18], and endothelial cells [19]. Necrotic cells also release HMGB1 into the extracellular milieu, where it induces the expression of several genes related to inflammation, including tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β), and leads to cell death [16,20]. In addition, this nuclear protein is endowed with extracellular signaling functions by interacting with different receptors such as the receptor for advanced glycation end products (RAGE) or toll-like receptor (TLR) 2 and TLR4 on the plasma membrane of various cell types [16,20].
RAGE, a transmembrane protein and a member of the immunoglobulin superfamily, is present on neurons, glia, and endothelial cells in the brain [21-24]. It interacts with various ligands, including HMGB1, advanced glycation end products (AGEs), β-amyloids, and S100 proteins [25]. In the cultured embryonic rat neurons, radiolabeled ligand binding studies with [125I] HMGB1 revealed that the binding affinity of HMGB1 to RAGE is seven-fold higher than HMGB1 to AGE, which was the first identified ligand for RAGE [26]. Interaction between RAGE and HMGB1 causes phosphorylation of mitogen-activated protein kinases (MAPKs; e.g., p38 and p42/44 kinases, stress-activated protein kinase/c-Jun N-terminal kinase, ERK1/2) and activation of the NF-kB signaling pathway in cultured macrophages, neutrophils, and Caco-2 epithelial cells [27,28]. This signaling is potentially linked to the receptor activation and is known to promote transcription of cytokines, such as TNF-α. Qiu et al. [29] found that HMGB1 treatment markedly increased the expression of the inflammatory mediators TNF-α, inducible nitric oxide synthase (iNOS) and intercellular adhesion molecule 1 (ICAM1) in the cultured glia and endothelial cells. They also reported the important participation of HMGB1 as a potential candidate in a specific upstream pathway promoting inflammation after brain ischemia [29]. Currently, the mechanism of interaction between HMGB1 and RAGE in the diabetic state is not well understood.
In the present study, we examined the relationship between the aggravation of cerebral ischemic injury following the stroke and the behavior of HMGB1 and its receptor, RAGE, in the diabetic rat brain after middle cerebral artery occlusion (MCAO) followed by reperfusion (MCAO/Re).
Materials and Methods
Experimental diabetic animals
Male Sprague Dawley rats (4 weeks old, weight 120�140g; Sankyo Labo Service Co., Ltd., Tokyo, Japan) were housed two to a cage in a temperature-controlled environment (23 � 0.5�C) with a 12- h light-dark cycle. The rats were given a standard rodent chow (CE-2; CLEA Japan, Inc., Tokyo, Japan) and water ad libitum. Animal care and surgical procedures were performed in accordance with the guidelines approved by the National Institutes of Health and the Josai University Animal Investigation Committee. A diabetic state was induced in the diabetic group (DM) rats by a single intraperitoneal injection of streptozotocin (STZ; 50mg/kg) dissolved in 0.1 mM sodium citrate (pH 4.5), while rats in the nondiabetic group (non-DM) were injected with buffer only [5]. Seven days after the STZ injection, a blood sample was collected by tail vein paracentesis and plasma glucose was determined using a glucose analyzer (Ascensia; Bayer Medical Co., Ltd., Land Nordrhein-Westfalen, Germany). Diabetes was defined as a blood glucose level >300mg/dl. The DM and non-DM groups were each divided into two groups (ischemic and control) and were housed for an additional 6 weeks until stroke was induced by MCAO (ischemic groups) or a sham operation was performed (control groups).
Middle cerebral artery occlusion and reperfusion
In the ischemic groups, MCAO was performed using a standard intraluminal procedure as previously described [5,30]. The rats were anesthetized with halothane (induction: 4%, maintenance: 1.5%) in 30% oxygen, using a face mask. Rectal temperature was maintained at 37�C with a heat lamp and a heating pad during the operation. A midline incision was made in the neck and the right common carotid artery was exfoliated under an operating microscope. All the branches of external carotid artery were ligated and isolated. The tips of 4-0 surgical nylon monofilaments were rounded by flame heating, and the rounded tips were inserted up through the internal carotid artery. Insertion was stopped when a small resistance was felt. Successful occlusion of the right middle carotid artery was confirmed when the left forelimb became paretic after the nylon filament was introduced. The distance from bifurcation of the common carotid artery to the tip of the suture was approximately 20 mm in all the rats. After occlusion for 2 h, the filament was withdrawn to allow for reperfusion. Cerebral blood flow was measured using Laser Doppler Flowmetry (ATBF-LC1; Unique Medical, Tokyo, Japan). There was an approximately 50% reduction in the baseline cerebral blood flow following MCAO. The animals were permitted to recover from the anesthesia at room temperature. The rats were sacrificed at 0.25, 0.5, 1, 3, 6, 12, 24, 48, and 72 h after reperfusion and the samples of brain and plasma were collected. The control groups underwent a sham operation with the same manipulation but without introduction of the monofilament.
Neurological evaluation
Postischemic neurological deficits were evaluated 2 h after MCAO, and 24 h after reperfusion using a 5-point scale as follows: grade 0, no deficit; grade 1, failure to extend right forepaw fully; grade 2, spontaneous circling or walking to the contralateral side; grade 3, walking only when stimulated; grade 4, unresponsive to stimulation and a depressed level of consciousness; and grade 5, death [5]. The rats that did not show neurological deficits were excluded from the study.
Infarct and edema assessment
At 0.25, 0.5, 1, 3, 6, 12, 24, 48, and 72 h after reperfusion, the rats were deeply anesthetized with halothane and decapitated. The brain was removed and cut into four 2mm coronal sections using a rat brain matrix, and was stained with 2% 2,3,5-triphenyltetrazolium chloride (TTC; Wako Pure Chemical Industries, Ltd., Osaka, Japan) at 37�C for 15 min. The coronal slices were fixed in 10% formaldehyde for photography. Infarct areas were determined using an image analysis system (Scion Image 1.62; Scion Corporation, Frederick, MD, USA), and were integrated to obtain the infarct volumes per brain. The total infarct volume was calculated by adding all cross-sectional areas multiplied by 2 mm (thickness of the sections). Correction of the infarct volume for edema was achieved using the following equation: corrected infarct volume (%) = [left hemisphere volume - (right hemisphere volume - the infarct volume)]/left hemisphere volume � 100. Edema in the ischemic hemisphere was also calculated: edema (%) = (right hemisphere volume - the infarct volume)/left hemisphere volume � 100 [5].
Evaluation of HMGB1 mRNA expression by real-time RT-PCR analysis
The rats subjected to MCAO were sacrificed at 1, 3, 6, 12, 24, 48, or 72 h after reperfusion. Brains were quickly removed, placed in RNA later (QIAGEN, Hilden, Germany) and stored at -80�C until further processing. A total RNA sample was obtained from the ischemic penumbral cortex of each rat. Analysis of the gene expression of HMGB1 was conducted using quantitative real-time reverse transcriptase polymerase chain reaction (RT-PCR) as previously described [10,11,31]. In brief, the total RNA was extracted from the ipsilateral cortex with an RNeasy Mini Kit (QIAGEN) according to the manufacturer�s instructions. Total RNA (0.5�g) from each sample was reverse-transcribed with oligo (dT) and random hexamer primers using reverse transcriptase (PrimeScript™ RT Enzyme Mix I, Takara RNA PCR Kit; Takara Biomedicals, Shiga, Japan). Real-time PCR was performed with 10 ng of cDNA and a pair of target gene specific primers (Takara Biomedicals) added to the SYBR Premix EX Taq (Takara Biomedicals) and subjected to PCR amplification in an iCycler iQ Real-Time Detection System (Bio-Rad Laboratories, Inc., Hercules, CA, USA; 1 cycle at 95�C for 10 s, and 50 cycles at 95�C for 5 s, and 60�C for 34 s). The expression of β-actin was used to normalize cDNA levels. The PCR products were analyzed by a melting curve to ascertain the specificity of amplification. The Primer sets were as follows: Hmgb1 (forward: GGAAATTAAAGCAGGAGGTTCTTGTTGG and reverse: CTGCATCAGAGACAACTGAAGATGG), β-actin (forward: GGAGATTACTGCCCTGGCTCCTA and reverse: GACTCATCGTACTCCTGCTTGCTG). The ratios of HMGB1 normalized to β-actin were considered as the expression of HMGB1 gene. All groups were compared to a nondiabetic-sham value of 1.
Immunohistochemistry
Immunohistochemical staining was performed as previously described [29,32]. In brief, the rats were sacrificed at the indicated time points and transcardially perfused with cold saline. Brains were fixed with 4% phosphate-buffered paraformaldehyde. Coronal brain sections (8�m thick) were incubated sequentially with 3% hydrogen peroxide at room temperature for 40 min to inhibit endogenous peroxidase, followed by incubation with blocking buffer (4% Block Ace; Dainippon Sumitomo Pharma Co. Ltd., Osaka, Japan) for 2 h. The slides were incubated with polyclonal rabbit anti-HMGB1 antibody (1:500, ab18256; Abcam Biotechnology, Cambridge, UK), polyclonal goat anti-RAGE antibody (1:200, sc-8230; Santa Cruz Biotechnology, CA, USA), monoclonal mouse anti-Neuronal nuclei (NeuN) antibody (1:500, MAB377; Chemicon, Temecula, CA, USA), monoclonal mouse anti-ionized calcium-binding adaptor molecule 1 (Iba1) antibody (1:500, ab15690; Abcam), or monoclonal mouse anti-glial fibrillary acidic protein (GFAP) antibody (1:500, 556327; BD Pharmingen, Franklin Lakes, NJ, USA) in 0.01 mol/l phosphate-buffered saline (PBS) at 4�C overnight. After washing with PBS, the slides were incubated with Cy3-conjugated donkey anti-rabbit IgG antibody (1:100, AP182C; Chemicon), Cy3-conjugated donkey anti-goat IgG antibody (1:100, AP180C; Chemicon) and FITC-conjugated goat anti-mouse IgG antibody (1:100, 81-6511; Zymed Laboratories, San Francisco, CA, USA) at room temperature for 2 h. Nuclei were stained with 4�,6-diamidino-2-phenylindole (DAPI; Invitrogen, Carlsbad, CA, USA) at room temperature for 15 min, washed, and mounted using mounting medium with 80% glycerol. Immunofluorescence was visualized using a Laser Scanning Confocal Microscope (Olympus Co. Ltd., FV10-ASW, Tokyo, Japan). Analysis was performed on nine photographs, three photographs for each experiment.
Western blot
Brain tissues, cerebrospinal fluid (CSF), and plasma were homogenized in SDS sample buffer [125mM Tris (pH 6.8), 4% SDS, 10% sucrose, 0.01% bromophenol blue, and 10% 2-mercaptoethanol] and boiled for 1 min. Protein concentration was quantified using the Bradford method (Protein Assay Reagent Kit; Bio-Rad Laboratories). The samples (40�g of protein from brain tissue and 10�l of CSF or plasma) were separated by SDS-polyacrylamide gel electrophoresis, transferred onto nitrocellulose membranes (Amersham Biosciences, Buckinghamshire, UK) through a semidry-type blotter (Bio-Rad Laboratories), blocked by 5% nonfat dry milk in PBS with Tween-20 (PBS-T; 137mM NaCl, 8.10mM Na2HPO4, 2.68mM KCl, 1.47mM KH2PO4, and 0.1% Tween-20), and incubated with appropriate antibodies as described below. The filters were incubated with each primary antibody at 4�C overnight and with the corresponding horseradish peroxidase (HRP)-conjugated secondary antibody in 5% nonfat dry milk/PBS-T at room temperature for 1 h. Finally, the target molecules were visualized through an enhanced chemiluminescence western blotting detection system (Amersham Biosciences) on X-ray film (Amersham Biosciences). Primary and secondary antibodies used in this study were polyclonal rabbit anti-HMGB1 (1:1,000, ab18256; Abcam), polyclonal goat anti-RAGE antibody (1:2,000, sc-8230; Santa Cruz), monoclonal mouse anti-β-actin (1:500, A2228; Sigma-Aldrich, St. Louis, MO, USA), HRP-conjugated goat anti-rabbit IgG antibody (1:5,000, NA934; Amersham Biosciences), HRP-conjugated donkey anti-goat IgG antibody (1:5,000, sc-2020; Santa Cruz), and HRP-conjugated sheep anti-mouse IgG antibody (1:10,000, NA931; Amersham Biosciences). Immunoblotted bands were quantified using Image Gauge software (Fujifilm, Tokyo, Japan) after densitometric scanning of the films.
Statistical analysis
Data are presented as means � S.D. Statistical analyses of mRNA levels and the area of cerebral damage were performed using a two-way ANOVA and post hoc Tukey�s multiple comparison tests. Neurological deficit scores were analyzed using the Kruskal�Wallis test followed by the Mann�Whitney U test. In all the cases, a P value of < 0.05 was taken as the level of significance.
Results
Postischemic neurological deficits were evaluated 2 h after MCAO and at various time points after reperfusion. There was no significant difference between non-DM (1.06 � 0.23) and DM (0.96 � 0.68) groups in the neurological deficit score 2 h after MCAO. However, after reperfusion, DM ischemic rats had significantly higher neurological deficit scores compared with those of non-DM ischemic rats. Death as a result of severe ischemic damage occurred in DM rats 48 h after reperfusion (Figure 1). There was no significant difference in regional cerebral blood flow among DM and non-DM ischemic groups and DM and non-DM controls (data not shown).
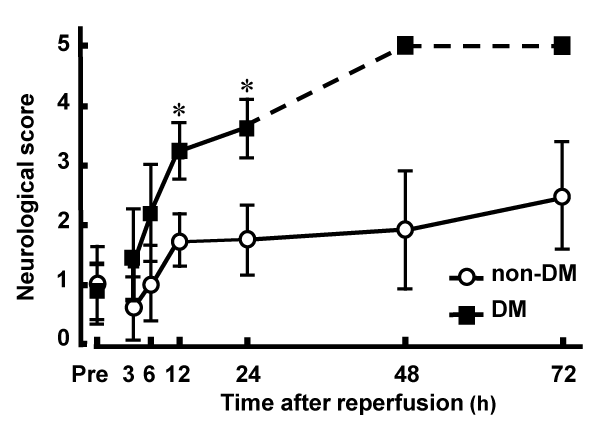
Figure 1: Neurological deficits induced by MCAO/Re in non-DM and DM
rats.
Postischemic neurological deficits were evaluated 2 h after MCAO (Pre)
and at various time points after reperfusion using a 5-point scale. The broken
line represents death of DM rats caused by severe ischemic damage 48 h
after reperfusion. Data are presented as means � SD (n=5�8 per time point).
*p< 0.01 vs. corresponding values for non-DM rats.
View Figure 1
Representative coronal brain sections of non-DM and DM ischemic rats at various time points after MCAO/Re were stained with TTC. Intact areas of the tissue stain with deep red originating from mitochondrial activity of the living cells, while infarcted tissue stains pale pink. The brain injury induced by MCAO/Re was considerably exacerbated by the diabetic state. In DM ischemic rats, the cerebral infarcts were produced within 30 min of reperfusion and the infarct regions extended to the whole thalamus during 12 h after reperfusion. In non-DM ischemic rats, only the small striatal infarcts were observed 12 h after reperfusion. The infarct volume in DM ischemic rats 6 h after reperfusion was significantly larger (about 10.8-fold) compared with that in non-DM ischemic rats (Figure 2A,2B). Brain edema was also exacerbated by the diabetic state; However, the difference between non-DM and DM ischemic rats was not significant (Figure 2A,2C).
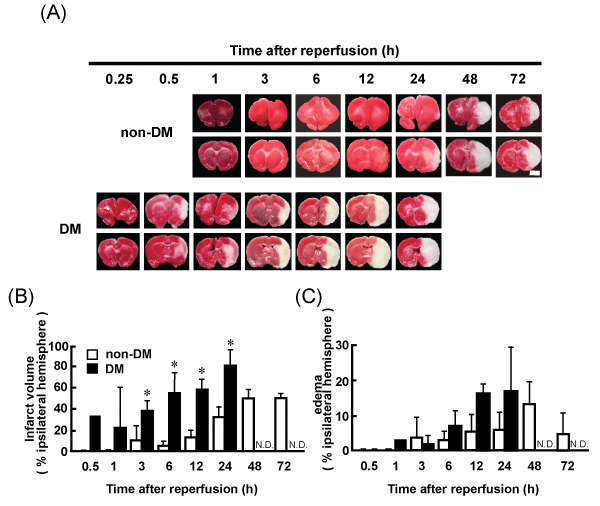
Figure 2: Brain infarct and edema in non-DM and DM ischemic rats detected by TTC staining
(A) Images of representative coronal brain sections from non-DM and DM rats stained by TTC at various time points after MCAO/Re showing viable (red) and
dead (pale pink) tissues. Scale bar=5mm. (B) Infarct volume (C) Edema in the ischemic hemispheres of non-DM and DM rats after MCAO/Re assessed by TTC
staining. Data are presented as means � SD (n=3�5 per time point). *p< 0.01 vs. corresponding values for non-DM rats.
View Figure 2
The level of HMGB1 mRNA in the total RNA from ischemic penumbral cortices was determined by real-time PCR. No apparent differences in the temporal expression pattern of HMGB1 were observed between non-DM and DM ischemic rats. HMGB1 gene expression was not affected by the cerebral ischemia during the 24 h after reperfusion, and was gradually reduced 48 h after reperfusion in the non-DM ischemic rats (Figure 3).
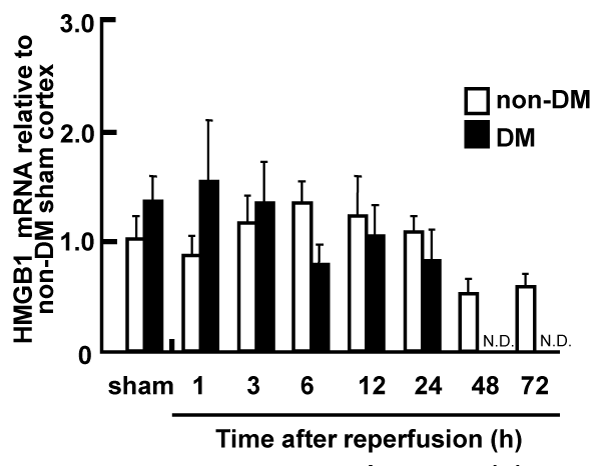
Figure 3: Expression of HMGB1 mRNA in the penumbral cortex of non-DM
and DM ischemic rats following MCAO/Re, relative to expression in shamoperated
non-DM rats
The expression level of HMGB1 mRNA was determined by real-time PCR
analysis in the penumbral cortex after MCAO/Re. Sham operation involved
the same manipulation but without insertion of the occlusive filament, as
described in Materials and Methods. Data are presented as means � SD
(n=3�4 per time point).
View Figure 3
Localization of HMGB1 in sham-operated (control) non-DM and DM rat cortical neurons was determined by immunohistochemical staining and confocal imaging. To determine whether neurons, microglia, and/or astrocytes express HMGB1, colocalization experiments with specific neuronal, microglial, and astrocytic markers were performed. HMGB1 (red) was widely expressed throughout the brain. Nuclear localization of HMGB1 was confirmed by counterstaining with DAPI (blue) and colocalized with NeuN (green). However, neither of the control groups showed colocalization with GFAP (green) or IBA1 (green). Translocation of HMGB1 from the nucleus to the cytoplasm was observed in DM but not in non-DM control rats (Figure 4A,4B). Therefore, we focused on the localization of HMGB1 in the nerve cells.
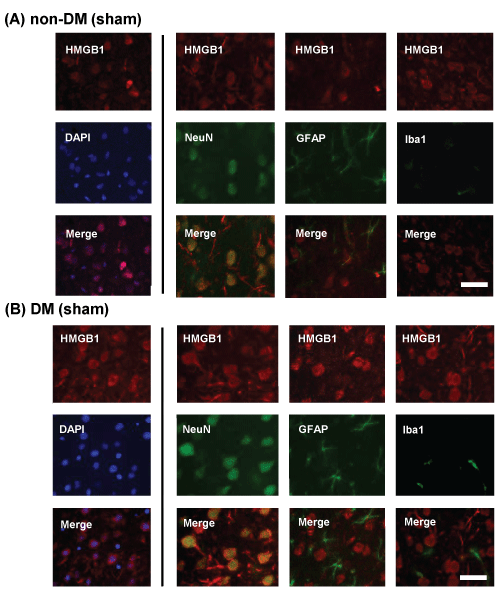
Figure 4: Immunohistochemical staining of HMGB1 in the penumbral cortex of sham-operated and ischemic non-DM and DM rats
Localization of HMGB1 was assessed using immunohistochemical staining of the penumbral cortex of (A) non-DM and (B) DM rats, 24 h after MCAO/Re.
Immunohistochemical staining of HMGB1 (red) was merged with DAPI (blue), NeuN (green), GFAP (green), and IBA1 (green). Sham-operation involved the same
manipulation but without the insertion of the occlusive filament, as described in Materials and Methods. Scale bar=20�m
View Figure 4
Translocation of HMGB1 from the nucleus to the cytoplasm in the neuronal cells induced by MCAO/Re in non-DM ischemic rats was detected during 6 or 12 h after reperfusion. A large amount of HMGB1 was observed in the cytoplasm even in sham-operated DM rats, and in DM ischemic rats. MCAO/Re accelerated the translocation of HMGB1 from the nucleus to the cytoplasm in the majority of the neurons from 3 h after reperfusion (Figure 5).
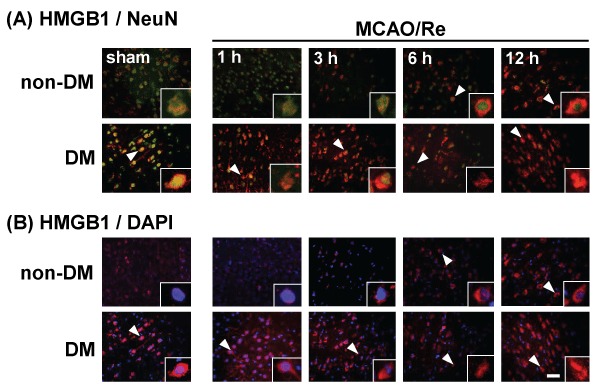
Figure 5: Translocation of HMGB1 in the penumbral cortex of sham-operated and ischemic non-DM and DM rats
Localization of HMGB1 was assessed using immunohistochemical staining of the penumbral cortex of non-DM and DM rats at various times after MCAO/Re.
Immunohistochemical staining of HMGB1 (red) was merged with (A) NeuN (green) or (B) DAPI (blue). Insets are higher magnification micrographs of the cells
indicated by arrowheads. Sham-operation involved the same manipulation but without insertion of the occlusive filament, as described in Materials and Methods.
Scale bar=20�m
View Figure 5
Immunohistochemical investigation showed a distinct border corresponding to the cortical peri-infarct region 1 h after MCAO/Re in DM ischemic rats. Immunoreactivity of HMGB1 disappeared in the core of the ischemic lesion after MCAO/Re in DM ischemic rats (Figure 6).
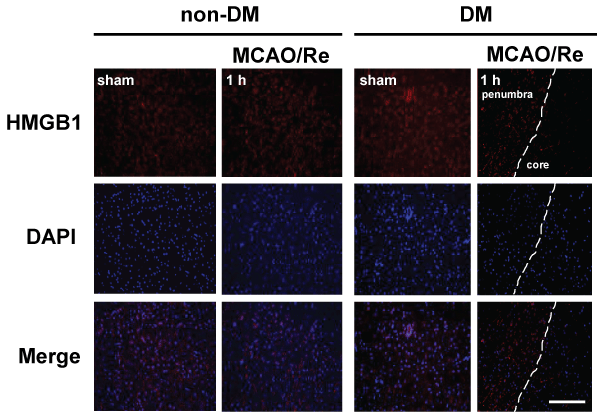
Figure 6: The early release of HMGB1 induced by MCAO/Re in DM ischemic rats
Localization of HMGB1 was assessed using immunohistochemical staining of the penumbral cortex of non-DM and DM ischemic rats 1 h after MCAO/Re.
Immunohistochemical staining of HMGB1 (red) was merged with DAPI (blue). The broken lines indicate the border between the core ischemic area (right side)
and the penumbra (left side). Sham-operation involved the same manipulation but without insertion of the occlusive filament, as described in Materials and
Methods. Scale bar=50�m
View Figure 6
HMGB1 levels in CSF and plasma after MCAO/Re were determined by the western blot analysis. No difference in the expression of HMGB1 was observed between the sham-operated non-DM and DM rats. The amount of HMGB1 in the CSF of non-DM ischemic rats increased gradually during 12 h after reperfusion. In contrast, HMGB1 in the CSF of DM ischemic rats was significantly increased approximately 3-fold 1 h after reperfusion, compared with after 12 h of reperfusion in non-DM ischemic rats (Figure 7A). Furthermore, HMGB1 levels in the plasma of DM ischemic rats showed a biphasic increase during 24 h after reperfusion. In the DM ischemic rats, plasma HMGB1 initially increased 1 h after reperfusion, by about 10-fold compared with non-DM ischemic rats. A second increase occurred up to 24 h after reperfusion, by about 3-fold compared with non-DM ischemic rats. The plasma HMGB1 in non-DM ischemic rats increased gradually during 24 h after reperfusion (Figure 7B).
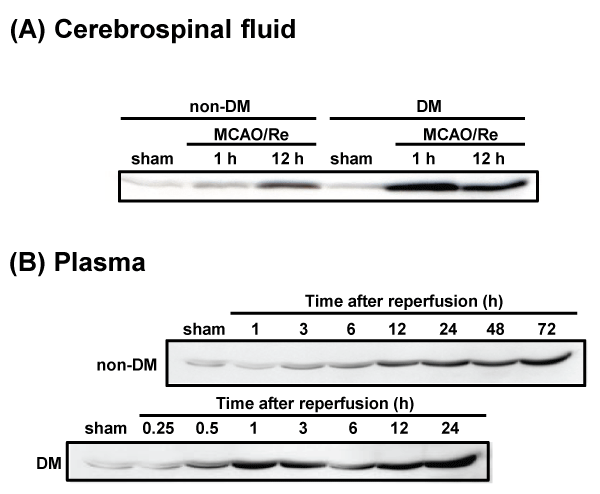
Figure 7: Temporal changes in cerebrospinal fluid and plasma HMGB1 after MCAO/Re or sham operation in non-DM and DM rats
HMGB1 in (A) CSF and (B) plasma was detected by western blotting at various times after MCAO/Re in non-DM and DM ischemic rats. Sham-operation involved
the same manipulation but without insertion of the occlusive filament, as described in Materials and Methods.
View Figure 7
The expression of RAGE in the rat brain was determined by immunohistochemical and western blot analyses. The immunoreactivity of RAGE gradually increased in the penumbral cortex of non-DM ischemic rats after MCAO/Re. RAGE expression was upregulated in sham-operated DM rats and increased up to 12 h after reperfusion in DM ischemic rats. Similar changes were also observed in the western blot analysis (Figure 8A-8C).
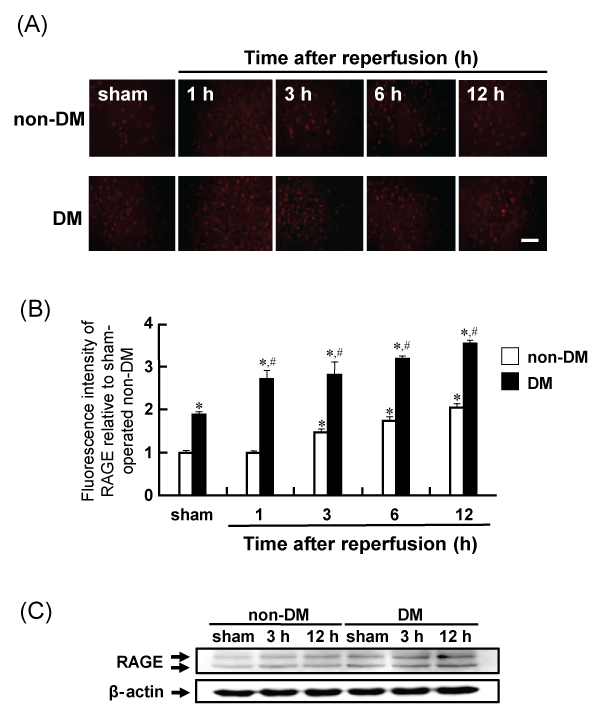
Figure 8: Levels of RAGE in the penumbral cortex after MCAO/Re or sham operation in non-DM and DM rats
(A) RAGE was assessed using immunohistochemical staining of the cortex of non-DM and DM rats at various times after MCAO/Re. (B) Quantitative analysis of
fluorescence intensity of RAGE in the penumbral cortex. (C) Western blot analysis of RAGE and β-actin from brain tissue was performed. Sham-operation involved
the same manipulation but without insertion of the occlusive filament, as described in Materials and Methods. Scale bar=50�m. Data are presented as means �
SD (n=5�8 per time point). *p< 0.01 compared with the non-DM sham group. #p< 0.01 compared with the DM-sham group.
View Figure 8
Discussion
The importance of inflammatory response in the pathophysiology of ischemic stroke is generally recognized. HMGB1 has been identified as an early mediator of hemorrhage after acute lung injury [33] and hepatic injury after liver ischemia/reperfusion [34]. Recent evidence suggests that the inflammatory cytokine HMGB1 is an active mediator of ischemic brain injury [29]. Moreover, elevated levels of HMGB1 in the serum of human patients with cerebral infarction have also been reported [35]. Therefore, HMGB1 has been attracting attention as a potential diagnostic marker [36,37].
HMGB1 was identified as a ubiquitously expressed, abundant nonhistone DNA-binding protein, which stabilizes the nucleosome formation and facilitates gene transcription. HMGB1 protein can be actively released into the extracellular space by macrophages and monocytes [16,20]. It is also passively released by necrotic cells, although not by apoptotic cells, and it triggers inflammation [38]. Extracellular HMGB1 interacts with different receptors, such as RAGE or TLR2/4 receptors, and promotes inflammatory responses leading to NF-κB activation [39,40]. Furthermore, HMGB1 has been reported to show delayed and sustained induction in the postischemic brain [41]. Previously, we revealed that the STZ-induced diabetic state aggravates cerebral ischemic injury following transient cerebral ischemia in the rats [5,10,11]. However, the mechanisms exacerbating the ischemic injury of the diabetic brain have not been elucidated. Therefore, in the present study, we examined the relationship between the aggravation of cerebral ischemic injury following the stroke in the diabetic state and the behavior of HMGB1 and its receptor, RAGE, in the rat brain after MCAO/Re.
The diabetic state aggravated MCAO/Re-induced neurological deficits and cerebral injury, as assessed by infarction volume. In the present study, the diabetic model had a significantly greater lesion volume and edema caused by MCAO/Re than previously reported [10,11]. Furthermore, we revealed that the infarct volume increases as early as 0.5 to 1 h after reperfusion in DM rats. HMGB1 gene expression was not affected by the cerebral ischemia in DM or non-DM rats. Immunohistochemical studies revealed that HMGB1 localized in the nuclei of the neuronal cells in sham-operated non-DM rat cortex, and was translocated from the nucleus to the cytoplasm of the neuronal cells during 6 or 12 h after reperfusion following MCAO in non-DM ischemic rats. We show that neurons are the principal sources of HMGB1 release in the early stages of ischemic injury. However, the translocation of HMGB1 was observed even in the sham-operated DM rat cortex. Furthermore, during MCAO/Re in DM rats, HMGB1 rapidly disappeared from all the cells within the cortex ischemic core from 1 h after reperfusion. Immunoreactivity against HMGB1 disappeared in the ischemic core and increased in CSF during 1 h after reperfusion in DM rat, suggesting earlier release of HMGB1 from necrotic neurons in the diabetic state.
Temporal changes in HMGB1 levels in CSF and plasma, an index of HMGB1 release, were also determined by immunoblot analysis. No difference in the amount of HMGB1 in CSF was observed between the sham-operated non-DM and DM rats. HMGB1 in the CSF of DM ischemic rats was considerably increased during 1 h after reperfusion, whereas in non-DM ischemic rats, HMGB1 in the CSF increased gradually during 12 h after reperfusion. The levels of HMGB1 in the plasma paralleled levels in the CSF. These results suggest that HMGB1 is readily translocated from the nucleus to the cytoplasm of the neuronal cells and immediately released after transient cerebral ischemia in the diabetic state. It has been reported that recombinant HMGB1 treatment increases inflammatory mediator gene expression in the cultured cells [29]. Therefore, the data reported herein suggest that HMGB1 may participate as an early upstream initiator of inflammation.
The expression of RAGE was upregulated in sham-operated DM rats and stabilized within 12 h after reperfusion in DM ischemic rats. The hyperglycemic conditions found in diabetes contribute to enhanced generation of AGE. Therefore, increases in expression of RAGE may contribute to generation of AGE and induce free radical generation [42]. Interestingly, genetic RAGE deficiency and decoy receptor soluble RAGE are reported to be associated with reduced infarct size [43].
HMGB1 causes activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and increased reactive oxygen species production in the neutrophils [44]. It also increases the expression of matrix metalloproteinase 9 (MMP9) and participates in the failure of the blood�brain barrier [45]. Therefore, it may contribute to the increased edema that is associated with the increased mortality from stroke [46]. Conceivably, increased expression of HMGB1 receptors after ischemia could enhance the reactivity of signaling by HMGB1-receptor interaction. [47-50]. Therefore, it is important to inhibit the HMGB1 released in response to ischemia during the treatment of postischemic injury in the diabetic patients.
In conclusion, the diabetic state induces pro-inflammatory cytokines in the brain, conceivably via hyperglycemia and/or oxidative stress, accelerating intracellular translocation and the release of HMGB1 from the neuronal cells after ischemic injury. An increase in extracellular HMGB1 may further induce inflammatory responses and cellular necrosis in the ischemic penumbra, leading to aggravation of ischemic injury in the diabetic state.
Acknowledgement
This study was supported by a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science to NI (No. 23790750). The authors would like to thank Enago (www.enago.jp) for the English language review.
Ethical Statement
All experiments were performed in compliance with the Guiding Principles for the Care and Use of Laboratory Animals approved by the Japanese Pharmacological Society, and the guidelines were approved by the Ethics Committee on Animal Care and Animal Experimentation at Josai University (#H26072). The number of animals used was kept to the minimum necessary for meaningful interpretation of the data. Animal discomfort was also minimized.
References
-
Stephens JW, Khanolkar MP, Bain SC (2009) The biological relevance and measurement of plasma markers of oxidative stress in diabetes and cardiovascular disease. Atherosclerosis 202: 321-329.
-
Baynes JW (1991) Role of oxidative stress in development of complications in diabetes. Diabetes 40: 405-412.
-
Vinik A, Flemmer M (2002) Diabetes and macrovascular disease. J Diabetes Complications 16: 235-245.
-
Biller J, Love BB (1993) Diabetes and stroke. Med Clin North Am 77: 95-110.
-
Iwata N, Okazaki M, Kasahara C, Kamiuchi S, Suzuki F, et al. (2008) Protective effects of a water-soluble extract from culture medium of Ganoderma lucidum mycelia against neuronal damage after cerebral ischemia/reperfusion in diabetic Rats. J Jpn Soc Nutr Food Sci 61: 119-127.
-
Rizk NN, Rafols J, Dunbar JC (2005) Cerebral ischemia induced apoptosis and necrosis in normal and diabetic rats. Brain Res 1053: 1-9.
-
Saito A, Maier CM, Narasimhan P, Nishi T, Song YS, et al. (2005) Oxidative stress and neuronal death/survival signaling in cerebral ischemia. Mol Neurobiol 31: 105-116.
-
Li PA, Liu GJ, He QP, Floyd RA, Siesj� BK (1999) Production of hydroxyl free radical by brain tissues in hyperglycemic rats subjected to transient forebrain ischemia. Free Radic Biol Med 27: 1033-1040.
-
Wei J, Quast MJ (1998) Effect of nitric oxide synthase inhibitor on a hyperglycemic rat model of reversible focal ischemia: detection of excitatory amino acids release and hydroxyl radical formation. Brain Res 791: 146-156.
-
Iwata N, Okazaki M, Kamiuchi S, Hibino Y (2010) Protective effects of oral administrated ascorbic acid against oxidative stress and neuronal damage after cerebral ischemia/reperfusion in diabetic rats. J Health Sci 56: 20-30.
-
Iwata N, Okazaki M, Xuan M, Kamiuchi S, Matsuzaki H, et al. (2014) Orally administrated ascorbic acid suppresses neuronal damage and modifies expression of genes encoding SVCT2 and GLUT1 in the brain of diabetic rats with cerebral ischemia�reperfusion. Nutrient 6: 1554-1577.
-
Javaherian K, Liu JF, Wang JC (1978) Nonhistone proteins HMG1 and HMG2 change the DNA helical structure. Science 199: 1345-1346.
-
Thomas JO (2001) HMG1 and 2: architectural DNA-binding proteins. Biochem Soc Trans 29: 395-401.
-
Rauvala H, Pihlaskari R (1987) Isolation and some characteristics of an adhesive factor of brain that enhances neurite outgrowth in central neurons. J Biol Chem 262: 16625-16635.
-
Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, et al. (1999) HMG-1 as a late mediator of endotoxin lethality in mice. Science 285: 248-251.
-
Erlandsson Harris H1, Andersson U (2004) Mini-review: The nuclear protein HMGB1 as a proinflammatory mediator. Eur J Immunol 34: 1503-1512.
-
Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, et al. (2003) Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J 22: 5551-5560.
-
Lotze MT, Tracey KJ (2005) High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol 5: 331-342.
-
Mullins GE, Sunden-Cullberg J, Johansson AS, Rouhiainen A, Erlandsson-Harris H, et al. (2004) Activation of human umbilical vein endothelial cells leads to relocation and release of high-mobility group box chromosomal protein 1. Scand J Immunol 60: 566-573.
-
Yang H, Wang H, Czura CJ, Tracey KJ (2005) The cytokine activity of HMGB1. J Leukoc Biol 78: 1-8.
-
Arancio O, Zhang HP, Chen X, Lin C, Trinchese F, et al. (2004) RAGE potentiates Abeta-induced perturbation of neuronal function in transgenic mice. EMBO J 23: 4096-4105.
-
Bierhaus A, Haslbeck KM, Humpert PM, Liliensiek B, Dehmer T, et al. (2004) Loss of pain perception in diabetes is dependent on a receptor of the immunoglobulin superfamily. J Clin Invest 114: 1741-1751.
-
Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, et al. (2003) RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med 9: 907-913.
-
Yan SD, Chen X, Fu J, Chen M, Zhu H, et al. (1996) RAGE and amyloid-beta peptide neurotoxicity in Alzheimer's disease. Nature 382: 685-691.
-
Huttunen HJ, Rauvala H (2004) Amphoterin as an extracellular regulator of cell motility: from discovery to disease. J Intern Med 255: 351-366.
-
Hori O, Brett J, Slattery T, Cao R, Zhang J, et al. (1995) The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J Biol Chem 270: 25752-25761.
-
Park JS, Arcaroli J, Yum HK, Yang H, Wang H, et al. (2003) Activation of gene expression in human neutrophils by high mobility group box 1 protein. Am J Physiol Cell Physiol 284: C870-879.
-
Huttunen HJ, Fages C, Kuja-Panula J, Ridley AJ, Rauvala H (2002) Receptor for advanced glycation end products-binding COOH-terminal motif of amphoterin inhibits invasive migration and metastasis. Cancer Res 62: 4805-4811.
-
Qiu J, Nishimura M, Wang Y, Sims JR, Qiu S, et al. (2008) Early release of HMGB-1 from neurons after the onset of brain ischemia. J Cereb Blood Flow Metab 28: 927-938.
-
Nakada Y, Yokoyama O, Komatsu K, Kodama K, Yotsuyanagi S, et al. (2000) Effects of aniracetam on bladder overactivity in rats with cerebral infarction. J Pharmacol Exp Ther 293: 921-928.
-
Liu K, Mori S, Takahashi HK, Tomono Y, Wake H, et al. (2007) Anti-high mobility group box 1 monoclonal antibody ameliorates brain infarction induced by transient ischemia in rats. FASEB J 21: 3904-3916.
-
Faraco G, Fossati S, Bianchi ME, Patrone M, Pedrazzi M, et al. (2007) High mobility group box 1 protein is released by neural cells upon different stresses and worsens ischemic neurodegeneration in vitro and in vivo. J Neurochem 103: 590-603.
-
Kim JY, Park JS, Strassheim D, Douglas I, Diaz del Valle F, et al. (2005) HMGB1 contributes to the development of acute lung injury after hemorrhage. Am J Physiol Lung Cell Mol Physiol 288: L958-965.
-
Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, et al. (2005) The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med 201: 1135-1143.
-
Goldstein RS, Gallowitsch-Puerta M, Yang L, Rosas-Ballina M, Huston JM, et al. (2006) Elevated high-mobility group box 1 levels in patients with cerebral and myocardial ischemia. Shock 25: 571-574.
-
Sapojnikova N, Kartvelishvili T2, Asatiani N2, Zinkevich V3, Kalandadze I4, et al. (2014) Correlation between MMP-9 and extracellular cytokine HMGB1 in prediction of human ischemic stroke outcome. Biochim Biophys Acta 1842: 1379-1384.
-
Oozawa S, Sano S, Nishibori M (2014) Usefulness of high mobility group box 1 protein as a plasma biomarker in patient with peripheral artery disease. Acta Med Okayama 68: 157-162.
-
Scaffidi P, Misteli T, Bianchi ME (2002) Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418: 191-195.
-
Yang QW, Wang JZ, Li JC, Zhou Y, Zhong Q, et al. (2010) High-mobility group protein box-1 and its relevance to cerebral ischemia. J Cereb Blood Flow Metab 30: 243-254.
-
Erlandsson Harris H, Andersson U (2004) Mini-review: The nuclear protein HMGB1 as a proinflammatory mediator. Eur J Immunol 34: 1503-1512.
-
Kim JB, Sig Choi J, Yu YM, Nam K, Piao CS, et al. (2006) HMGB1, a novel cytokine-like mediator linking acute neuronal death and delayed neuroinflammation in the postischemic brain. J Neurosci 26: 6413-6421.
-
Prasad S, Sajja RK1, Naik P1, Cucullo L2 (2014) Diabetes Mellitus and Blood-Brain Barrier Dysfunction: An Overview. J Pharmacovigil 2: 125.
-
Muhammad S, Barakat W, Stoyanov S, Murikinati S, Yang H, et al. (2008) The HMGB1 receptor RAGE mediates ischemic brain damage. J Neurosci 28: 12023-12031.
-
Tang D, Kang R, Zeh HJ 3rd, Lotze MT (2011) High-mobility group box 1, oxidative stress, and disease. Antioxid Redox Signal 14: 1315-1335.
-
Qiu J, Xu J, Zheng Y, Wei Y, Zhu X, et al. (2010) High-mobility group box 1 promotes metalloproteinase-9 upregulation through Toll-like receptor 4 after cerebral ischemia. Stroke 41: 2077-2082.
-
Xiong XX, Gu LJ, Shen J, Kang XH, Zheng YY, et al. (2014) Probenecid protects against transient focal cerebral ischemic injury by inhibiting HMGB1 release and attenuating AQP4 expression in mice. Neurochem Res 39: 216-224.
-
Hu X, Cui B, Zhou X, Xu C, Lu Z, et al. (2012) Ethyl pyruvate reduces myocardial ischemia and reperfusion injury by inhibiting high mobility group box 1 protein in rats. Mol Biol Rep 39: 227-231.
-
Su X, Wang H, Zhao J, Pan H, Mao L (2011) Beneficial effects of ethyl pyruvate through inhibiting high-mobility group box 1 expression and TLR4/NF-κB pathway after traumatic brain injury in the rat. Mediators Inflamm 2011: 807142.
-
Zhang J, Takahashi HK, Liu K, Wake H, Liu R, et al. (2011) Anti-high mobility group box-1 monoclonal antibody protects the blood-brain barrier from ischemia-induced disruption in rats. Stroke 42: 1420-1428.
-
Liu K, Mori S, Takahashi HK, Tomono Y, Wake H, et al. (2007) Anti-high mobility group box 1 monoclonal antibody ameliorates brain infarction induced by transient ischemia in rats. FASEB J 21: 3904-3916.
