

International Journal of Diabetes and Clinical Research
Involvement of Telmisartan on the Protective Effects Mediated by the Peroxisome Proliferator-Activated Receptor-γ Pathway in Mice
Chenghu Huang, Li Yuan* and Shuyi Cao
Department of Endocrinology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, China
*Corresponding author:
Li Yuan, M.D., Ph.D., Department of Endocrinology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430022, China, Tel: +86-027-85726300; E-mail: yuanli18cn@163.com
Int J Diabetes Clin Res, IJDCR-1-015, (Volume 1, Issue 3), Review Article; ISSN: 2377-3634
Received: November 12, 2014 | Accepted: December 04, 2014 | Published: December 08, 2014
Citation: Huang C, Yuan L, Cao S (2014) Involvement of Telmisartan on the Protective Effects Mediated by the Peroxisome Proliferator-Activated Receptor-γ Pathway in Mice. Int J Diabetes Clin Res 1:015. 10.23937/2377-3634/1410015
Copyright: © 2014 Huang C, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Aims: The aim of this study is to further elaborate the improving effects of telmisartan on insulin resistance, beta-cell dysfunction and islet inflammation in a PPAR-γ signaling-dependent manner.
Methods: The mRNA levels of renin-angiotensin system components were detected. We compared the effects of telmisartan and telmisartan combined with GW9662 (a PPAR-γ antagonist) in mice fed a high-fat diet or isolated non-diabetic mouse islets. Immunofluorescence or real-time PCR was used to determine changes in islet structure, function and inflammation as well as PPAR-γ expression.
Results: Telmisartan reduced body weight, especially the visceral adipose tissue weight, which may be due to enhancing PPAR-γ expression and reshaping the RAS balance, including increased ACE2 and decreased ACE/ATR1 mRNA expression levels. IPGTT and HOMA-IR showed that telmisartan attenuated insulin resistance in a manner dependent on the duration of high-fat diet fed, which could be offset by GW9662. In addition, telmisartan normalized beta-cell function (including the increase in PDX1, GLUT2 and insulin mRNA expression levels) and islet structure (such as the maintenance of islet cell distribution, decrease in the alpha/beta ratio and alpha-cell mass and the inhibition of beta-cell apoptosis) via the activation of PPAR-γ signaling in mice fed a high-fat diet. Moreover, telmisartan inhibited the high fat diet-induced activation of p65 expression and inflammatory factors (including IL-1beta and TNF-alpha) which were stimulated by chronic palmitate exposure in pancreatic islets, while GW9662 had the reverse effect.
Discussions: Our novel results exhibit critical roles for PPARγ activation in the biological effects of telmisartan and suggest a therapeutic potential for telmisartan in the prevention of type 2 diabetes.
Keywords
Telmisartan, PPAR-γ, Insulin resistance, Beta-cell dysfunction, Inflammation
Introduction
The primary characteristics of type 2 diabetes are insulin resistance, relative insulin deficiency and frequent hyperglycemia. Interestingly, several lines of evidence have implicated the renin-angiotensin system (RAS), especially angiotensin II, as a key factor in the initiation and progression of these disorders [1]. The RAS consists primarily of an enzymatic cascade through which angiotensinogen is converted to angiotensin I (Ang I), which is then converted to Ang II by angiotensin-converting enzyme (ACE). Most of these functions are mediated by the type 1 receptor (AT1R), which is balanced by the 'new' arm of the RAS, namely the ACE2/Ang-(1-7)/Mas receptor axis [2,3]. Inhibition of the RAS via ACE inhibitors (ACEI) and AT1R blockers (ARB) consistently and significantly decreases the new onset of type 2 diabetes [4,5]. The probable mechanism is pleiotropic. The protective effects are mainly due to reduced oxidative stress, inflammation and pro-inflammatory cytokine secretion [6].
Unlike other ARBs, telmisartan is a partial agonist of peroxisome proliferator activated receptor-γ (PPAR-γ); this property is implicated in improving insulin sensitivity by reducing adipocyte size and modulating the inflammatory state of adipose tissue [7-9]. PPAR-γ is a major regulator of lipid, glucose, and amino acid metabolism [10]. Yamana A et al. reported that telmisartan has beneficial effects on insulin sensitivity and glycemic control in hypertensive patients [11]. In addition, a recent study showed that telmisartan protects pancreatic ultrastructural enhancement due to activation of PPAR-γ [12]. Nonetheless, the protective effect of telmisartan mediated by the PPAR-γ pathway is not completely understood. In the present study, we again confirmed that telmisartan reshaped the RAS balance and show that the elevated activation of PPAR-γ stimulated by telmisartan was beneficial to the promotion of insulin resistance, the normalization of islet structure, the preservation of islet cell mass and the inhibition of intra-islet inflammation, which has been attributed to abrogation of the detrimental effects exerted by high-fat diets.
Material and Methods
Animals
The mice were maintained under standard light conditions (12 h light, 12 h dark cycle) and allowed free access to water and food. Male C57BL/6 mice and food were purchased from The Beijing HFK Bio-Technology. Co., Ltd. Beijing, China. The care and experimental treatment of the animals were approved by the Animal Research Committee of Tongji Medical College, Huazhong University of Science and Technology. Six-week-old mice weighing 21 � 2 g were randomized to either a vehicle-treated (0.5% carboxymethyl cellulose) or telmisartan-treated (5mg�kg-1�d-1) group (all applied by oral gavage) (Sigma-Aldrich, MO, US) and fed with a high-fat diet (60% kcal from fat). Until the mice were 10 weeks old, the telmisartan-treated group was divided into the treatment with telmisartan plus GW9662 (GW9962, a PPAR-γ antagonist, 10mg�kg-1�d-1) (Sigma-Aldrich, St. Louis, MO, US) and telmisartan alone (telmisartan) groups. Age-matched low-fat diet (LFD (10%kcal from fat)-fed and vehicle-treated mice served as controls. Body weight and food intake were measured weekly. To exclude differences induced by food intake, all high-fat diet-fed mice were pair-fed. This food scheme lasted 8 weeks and aimed to enhance the development of metabolic syndrome features. After 8 weeks of drug treatment, mice were anesthetized with ether, blood was sampled by cardiac puncture, and plasma samples were collected by centrifugation and stored at -80�C until use.
Islet isolation and cell culture
Non-diabetic mouse islets were isolated from pancrease according to previously described methods [13]. The pancreas was perfused through the common bile duct with 1.5 mg�mL-1 collagenase P (Roche Applied Science, Indianapolis, IN, US), incubated at room temperature, and then further separated from the acinar tissue using a Histopaque 1077 gradient. The islets were isolated by hand and cultured for 24�48 h in a 5% CO2 incubator in RPMI 1640 medium (3mmol�L-1 glucose) supplemented with glutamine (2mmol�L-1), 10% (v/v) heat-inactivated fetal bovine serum (Gibco, Gaithersburg, MD, US) and antibiotics. Then the islets assessed according to the steps outlined in functional studies.
Functional studies
Non-diabetic islets of 6-week mice were seeded into 24-well plates, and 25 islets were added to each well. The palmitate solution was prepared as previously described [14]. After incubation in KRB buffer with 5.6mmol�L-1 glucose for 1 h, the islets were incubated under the following conditions: 0.5% BSA (as a control) or 0.5mmol�L-1 palmitate bound to 0.5% BSA for 48 h. Telmisartan (10mmol�L-1) were added into the wells in the presence of 0.5mmol�L-1 palmitate in separate experiments. GW9662 (300nmol�L-1) was added in addition to 10mmol�L-1 telmisartan in separate experiments. The plate was gently shaken to allow thorough mixing of drugs before the non-diabetic mouse islets were transferred to the wells for 48 h incubation at 37�C.
Metabolic measurements
Two tests were performed in both of the mouse groups, the first after 4 wks on a high-fat diet and the second after 8 wks on a high-fat diet. For intraperitoneal glucose tolerance tests (IPGTT), mice were fasted overnight and intraperitoneally (ip) injected with 2g�kg-1 glucose the following morning. Blood glucose was measured with an UltraTouch glucometer from cut tail tips at 0, 30, 60 and 120 min following glucose injection. For the fasting glucose and insulin, mice were fasted 6 h. The insulin concentration was determined by the insulin ELISA kit (Millipore, Billerica, MA, US). Therefore, we calculated HOMA-IR (Homeostasis model for assessment of insulin resistance, fasting glucose (mmol�L-1) * fasting insulin (mU�L-1)/22.5).
Immunodetection
Paraffin sections (5�m thick) were rehydrated, and antigen retrieval was performed using a PickCell pressure cooker. The primary antibodies used were guinea pig anti-insulin (1:150; Abcam, Cambridge, UK), rabbit anti-glucagon (1:200; Cell Signaling, Denver, MA, US), rabbit anti-active Caspase-3 (1:100; Abcam) and rabbit anti-p65 (1:200; Abcam) and rabbit anti-PPAR-γ (1:150; Abcam). Secondary antibodies conjugated with Alexa Fluor 488 (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) and Dylight 549 (Abbkine, CA, USA) was used. The nuclear counterstain 4�6�-diamidino -2-phenylindole (DAPI, 1:1000; Invitrogen, Carlsbad, CA, US) was also used. Then, border of a pancreatic section was visually defined on a composite picture of entire section displayed on a computer screen. The digital images were acquired by a fldigital im microscope equipped with a DC 200 digital camera (C-1/TE200U; Nikon, Tokyo, Japan) and were then analyzed with the Image-Pro Plus software, version 5.0 (Media Cybernetics). The density threshold selection tool was used to select areas of the pancreatic islet marked with insulin and glucagon, which were then expressed as percentages of the islet mean cross-sectional area (immuno-density) [15,16]. The beta-cell area (%) was calculated as (sum of beta cell area/total pancreas area). Beta-cell mass was calculated as (weight of prefixed pancreas) � (beta-cell area/total pancreas area). Similarly, alpha-cell mass and cleaved caspase-3 positive cell mass were also calculated.
mRNA expression Analyses
Total RNA was extracted from isolated islets, mouse pancreatic tissues and epididymal white adipose tissue using Trizol reagent (Invitrogen). The generation of first-strand cDNA was achieved using a cDNA synthesis kit (Takara Shuzo, Kyoto, Japan). Real-time polymerase chain reaction (Q-PCR) was performed using a LightCycler (Roche Diagnostics GmbH, Mannheim, Germany). Relative transcript levels were normalized to those of 36B4 and calculated using the 2-ΔΔCT statistical method. The primers used in this experiment are listed in Table 1.
![]()
Table 1: Effect of the respective diets on body weight and serum glucose (n = 7).
View Table 1
Statistical analysis
Results are expressed as the mean � SEM. Differences between groups were carried out using two-tailed unpaired Student�s t test or one-way ANOVA followed by post hoc comparisons using Tukey corrections. P-values < 0.05 were considered statistically significant.
Results
Mice assessment
After being fed for 3 weeks on their respective diets, the HFD group started to present higher BWs, while the telmisartan group began to show a resisting effect against the high-fat diet. After 5 weeks, GW9662 expeditiously abrogated the effect of telmisartan and finally resulted in a significantly increased BW (+18.39%) (Figure 1A). After eight weeks of diet consumption, the BW of HFD group markedly increased (+20.07%) compared with the LFD group while telmisartan decreased the BW of HFD group (-12.87%) (Figure 1A).
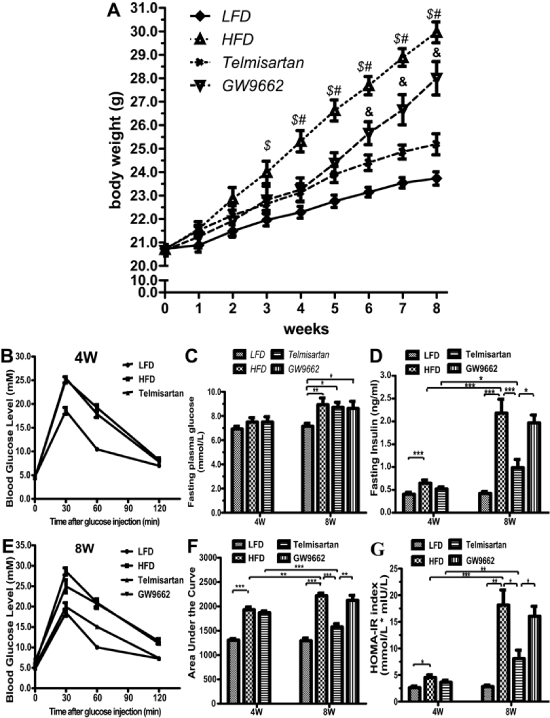
Figure 1: Body weight evolution, glucose metabolism and hormone levels between groups on their respective diets for 8 weeks. A. Body weight evolution in each group. LFD: Low-Fat Diet; HFD: High-Fat Diet alone; Telmisartan: high-fat diet and telmisartan treatment; GW9662: high-fat diet and telmisartan treatment plus GW9662 administration for 4 weeks. n=7 mice per group; $ P < 0.05 LFD vs. HFD group; # P < 0.05 HFD vs. Telmisartan group; & P < 0.05 Telmisartan vs. GW9662 group. B-C, Serum fasting glucose (B) and insulin levels (C) were determined in 6-hour-fasted mice of each groups (4 and 8 wk). D-F. Intraperitoneal glucose and insulin tolerance tests (IPGTTs) and area under curve (AUC, #210) analysis of the mice. Blood glucose levels were measured from cut tail tips using an UltraTouch glucometer at 0 min, 30 min, 60 min and 120 min after glucose injection. I, The homeostatic model assessment of insulin resistance (HOMA-IR) index. HOMA-IR = fasting glucose (mmol�L-1) * fasting insulin (mU�L-1)/22.5. Values are the mean � SEM. LFD: low-fat diet; HFD: high-fat diet alone; Telmisartan: high-fat diet and telmisartan treatment; GW9662: high-fat diet and telmisartan treatment plus GW9662 administration for 4 weeks. n=7 mice per group; $ P < 0.05 LFD vs. HFD group; # P< 0.05 HFD vs. Telmisartan group; & P< 0.05 Telmisartan vs. GW9662 group;*P< 0.05; **P< 0.01; ***P< 0.001.
View Figure 1
In addition to the palpable difference in BW, the effect of telmisartan and GW9662 on adipose tissue weight was equivocal. HFD mice exhibited a nearly 2.0-fold increase in total adipose tissue weight, compared with LFD mice (Table 2). This was attributed to fat accumulation in subcutaneous, perirenal, and epididymal depots (P< 0.01 for all depots). At the end of the experiment, marked protection against the high-fat diet resulted from treatment with telmisartan (58.73% decrease in total adipose tissue weight); however, this protection was reversed by treatment with GW9662 (6.67% increase). Intriguingly, a similar effect was shown on liver weight with the respective diets (for all depots) (Table 2).
![]()
Table 2: List of primers used for quantitative PCR using SYBR Green.
View Table 2
Glucose metabolism and hormone levels
The high-fat diet increased fasting plasma glucose levels at 8 weeks, but not at 4 weeks (Figure 1B). However, the fasting insulin levels markedly increased at 4 weeks, and the tread is increasingly greater at 8 weeks (Figure 1C). As expected, telmisartan decreased the elevated fasting insulin level induced by high-fat diet, and GW9662 had a reverse effect (Figure 1C). Compared with the IPGTT at 4 weeks, telmisartan ameliorated glucose tolerance based on the lower concentrations of insulin necessary to equilibrate the plasma glucose at 8 weeks. The difference between the HFD and telmisartan groups was not significant at 4 weeks, but the difference was unequivocal at 8 weeks (Figure 1D,1F). GW9662 abrogated the effect of telmisartan on IPGTT (Figure 1D,1F). Furthermore, telmisartan improved insulin sensitivity, and GW9662 exacerbated insulin resistance, as shown by the HOMA-IR, supporting the fact that telmisartan exerts metabolic effects through the activation of PPAR-γ (Figure 1G).
The expression of renin-angiotensin system components
The high-fat diet disrupted the RAS balance in epididymal white adipose tissue (EWAT), including the increase in ACE and ATR1 mRNA expression levels and the decrease in ACE2 mRNA levels (Figure 2A-C). Telmisartan, as a classical ATR1 blocker, significantly lessened ATR1 mRNA expression levels (Figure 2B). Importantly, telmisartan also reshaped the balance by elevating ACE2 and reducing ACE expression levels (Figure 2A,2C), and in turn the ACE/ACE2 ratio decreased to an even lower extent (Figure 2E). Consistent with the expression of ACE2 mRNA expression, the expression of Ang (1-7) receptor MAS mRNA was lessened by high-fat diet and induced by telmisartan (Figure 2D). Because endocrine pancreatic tissue harbors a full complement of the RAS [1], we also explored the effects of lipotoxicity in isolated non-diabetes mouse islets. Intriguingly, the expression of the RAS components accomplished the same result in the islets. Prolonged palmitate exposure induced the increase of ACE (Figure 2F) and ATR1 mRNA (Figure 2G) expression, and the decrease of ACE2 (Fig. 2H) and MAS mRNA (Figure 2I) expression, which could be abrogated by telmisartan (Figure 2F-I). Certainly, ACE/ACE2 ratio in local pancreatic islet was normalized by telmisartan (Figure 2J). Thus, telmisartan indeed reshaped the balance of local RAS both in pancreatic islet and white adipose tissue.
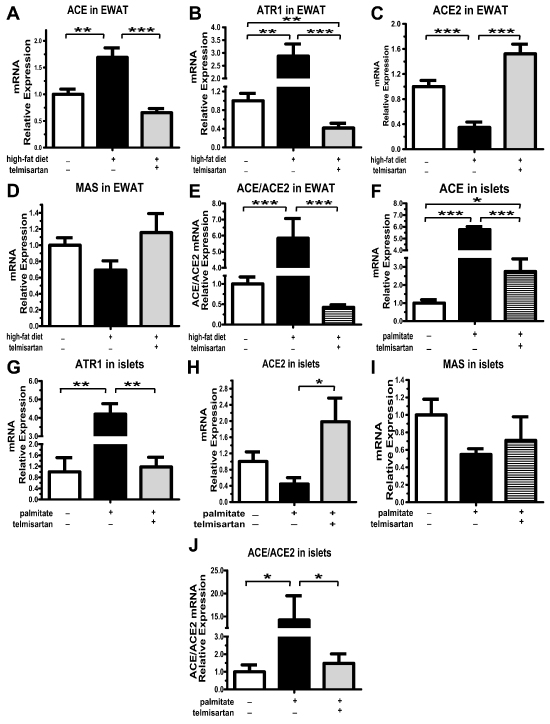
Figure 2: mRNA expression levels of RAS components in epididymal white adipose tissue and isolated non-diabetic mouse islets. After mice on their respective diets for 8 weeks, mRNA expression of ACE (A), ATR1 (B), ACE2 (C) and MAS (D) in the epididymal white adipose tissue (EWAT) detected by Q-PCR. E. ACE/ACE2 relative mRNA expression levels in the EWAT. n=7 mice per group. After incubation isolated non-diabetic mouse islets with 0.5 mmol�L-1 palmitate for 48 h, mRNA expression of ACE (F), ATR1 (G), ACE2 (H) and MAS (I) were detected by Q-PCR. E. ACE/ACE2 relative mRNA expression levels in the islets. Results are normalized via the reference gene 36B4. n=3 separate isolated islet preparations; *P< 0.05; **P< 0.01; ***P< 0.001.
View Figure 2
The PPAR-γ expression
As expected, immunofluorescence showed that the expression of PPAR-γ protein was mainly localized in pancreatic islets (Figure 3A). The high-fat diet significantly inhibited PPAR-γ expression levels, which was reversed by telmisartan (Figure 3A). Meanwhile, the high-fat diet led PPAR-γ mRNA levels to drop to less than half. Although telmisartan offset this drop, the levels of PPAR-γ mRNA in the telmisartan group were still lower than those in the LFD group (Figure 3B). PPAR-γ mRNA levels in epididymal fat followed the same trend, which seemed to be more marked than in the pancreas (Figure 3B-C). We would like to stress, however, that GW9662 inhibited PPAR-γ expression at both the mRNA and protein levels (Figure 3A-C).
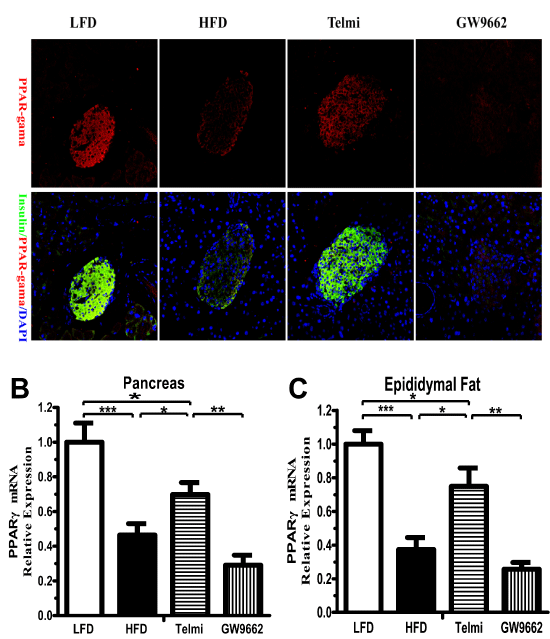
Figure 3: PPAR-γ expression in adipose tissue and the pancreas. A, Representative images of immunofluorescence staining for insulin (green, #59) and PPAR-γ (red). PPAR-γ mRNA expression levels were detected in the pancreas (B) and epididymal white adipose tissue (C). Normalization of the results with an endogenous standard was performed via the reference gene 36B4.HFD: high-fat diet alone; Telmi: telmisartan + HFD group; GW9662 (telmisartan + HFD plus GW9662) group. n=7 mice per group; *P< 0.05; **P< 0.01; ***P< 0.001.
View Figure 3
Beta-cell mass, function, apoptosis and islet architecture
Immunofluorescence revealed that mice fed a high-fat diet maintained intact islets based on the islet architecture of insulin and glucagon. Unlike the defined alpha-cell mantle and beta-cell core characteristics of LFD islets, HFD islets maintained a more scattered organization and a higher percentage of alpha-cells. The increase in alpha-cell mass did not occur with telmisartan treatment, and in this group, the alpha/beta ratio was evidently lower, which likely could be attributed to beta-cell hypertrophy induced by the high-fat diet (Figure 4A,4B). In accordance with the effects on the alpha/beta ratio, the HFD and GW9662 groups exhibited a significant increase in alpha-cell mass (Figure 4B,4D). The differences in islet size and beta-cell mass between the LFD and HFD groups were marked; however, the differences were not notable between the two HFD groups (Figure 4C). Immunofluorescence revealed a marked decrease in cleaved caspase-3 in telmisartan-treated mice on a high-fat diet (Figure 4C,4E). The gene expression levels of beta-cell markers, such as insulin, PDX-1 and GLUT2, at least at the mRNA level, were lower in the HFD group than in the LFD group; this effect was reversed by telmisartan (Figure 4G).
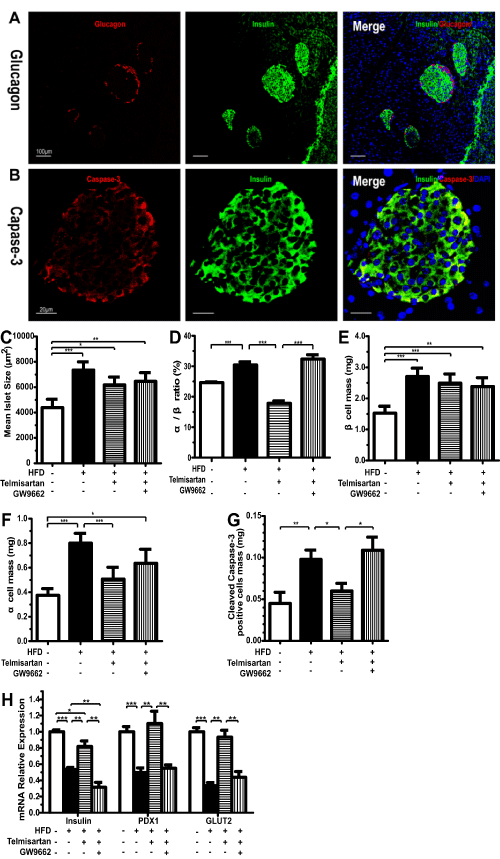
Figure 4: Beta-cell mass, functions, apoptosis and islet architecture. A, Representative images of immunofluorescence staining for insulin and glucagon (�100) or Cleaved Caspase-3 and insulin (�400) of mice on their respective diets for 8 weeks. Representative images of staining for insulin (green, #59) and glucagon (red) (A) or active caspase-3 (red) (B). After immunofluorescence, areas labeled for insulin (beta-cell) and glucagon (alpha-cell) were measured, and the results are expressed as the mean islet size (C), alpha/beta ratio (D), beta-cell mass (E) and alpha-cell mass (F). G, mRNA expression levels of insulin, PDX1 and GLUT2 detected by Q-PCR in pancreas of mice on their respective diets for 8 weeks. n=7 mice per group; *P< 0.05; **P< 0.01; ***P< 0.001.
View Figure 4
NF-kB inflammatory signaling pathway
Immunofluorescence showed that p65 was held in the cytoplasm in an inactive state in the LFD group and that the high-fat diet induced increased expression and translocation of p65 to the nucleus. Telmisartan decreased the effects of the high-fat diet, and the effect of telmisartan could be abrogated by treatment with GW9662 (Figure 5A). We also investigated the target of NF-κB pathway, including TNF-alpha and IL-1beta mRNA expression. The mRNA expression of IL-1beta and TNF-alpha markedly increased by 4.70- and 3.62-fold, respectively (Figure 5B-C). Telmisartan significantly inhibited the elevated trend induced by palmitate, however, the expression of TNF-alpha mRNA was higher than control (p=0.44) (Figure 5B,5C). Unequivocal, GW9662 could thoroughly eliminate the inhibition of telmisartan and exacerbated the effects of palmitate overload on the expression of TNF-alpha mRNA (p=0.52 and 0.24, respectively) (Figure 5B,5C).
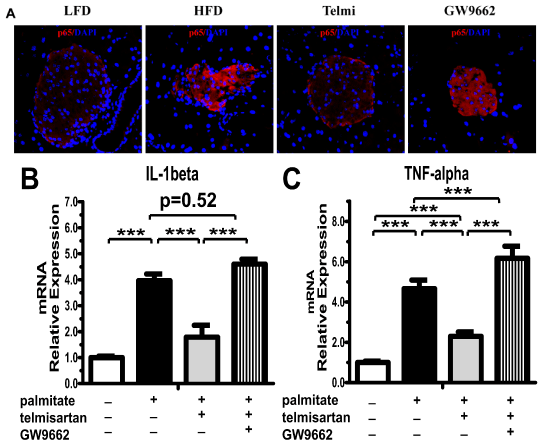
Figure 5: Telmisartan decreased NF-κB protein expression via upregulation of the PPAR-γ pathway. A. Representative images of immunofluorescence staining for p65 (�400) in the LFD, HFD, Telmi (telmisartan + HFD) or GW9662 (telmisartan + HFD plus GW9662) group. n=7 mice per group. After pre-incubation with 100nmol�L-1 liraglutide for 2h, followed by exposure to 0.5mmol�L-1 palmitate for 48 h, IL-1beta (B) and TNF-alpha (C) mRNA were detected by Q-PCR in isolated non-diabetic mouse islets. n=3 separate isolated islet preparations. *P< 0.05; **P< 0.01; ***P< 0.001.
View Figure 5
Discussions
The present study rationalizes the idea that telmisartan abrogates high-fat diet-induced insulin resistance and beta cell dysfunction in a time-dependent and PPAR-γ-dependent manner. As expected, this protection included the decrease in total adipose tissue weight, the maintenance of normal islet structure, the inhibition of beta-cell apoptosis and the suppression of islet inflammation. In addition to remodeling the RAS balance, activation of the PPAR-γ pathway may be an alternative molecular mechanism to explain the protective effects of telmisartan, as the PPAR-γ antagonist GW9662 partially suppressed the protective effects of telmisartan.
Recently, the classical concept of the RAS has undergone substantial conceptual changes due to the identification of the ACE2/Ang (1-7)/Mas axis, which is an endogenous modulator of ACE/Ang II/ATR1 signaling. Because Ang II is generated by ACE, and ACE2 cleaves Ang II to Ang (1-7), the unusual RAS balance switches between the generation of Ang II and Ang (1-7) depending on the relative levels of ACE and ACE2, respectively [2,17]. Interestingly, the activation of Ang II/AT1R signaling is upstream of ACE and lessens ACE2 expression via the p38 MAPK and ERK1/2 pathways [17,18]. Our data demonstrated that telmisartan, an AT1R blocker, could reshape the local balance of the RAS, including increased ACE2 and decreased ACE/ATR1 mRNA expression levels in epididymal white adipose tissue and pancreatic islet. Concurrent with our recent findings there are pulications that demonstrating that ACEs or ARBs not only attenuated insulin resistance but also normalized islet structure and beta-cell function and inhibited beta-cell apoptosis [5,19,20], which arises both from the circulating endocrine system and the local tissue paracrine system. Particularly, in adipose tissue Ang II plays a vital role in adipose cell differentiation, lipid accumulation and tissue dysfunction [21,22]. In local pancreatic islets, concurrent with our previous findings there are publications that the RAS also regulates islet function and glycemic control via influences on islet cell mass, inflammation, and ion channels [23,24].
PPAR-γ signaling is an alternative molecular mechanism that mediates the protective effects of telmisartan. Our data showed that PPAR-γ mediated the inhibitory regulatory mechanism of telmisartan in a complicated and pleiotropic manner. First, PPAR-γ signaling contributed to body weight loss, especially to the decrease in visceral fat accumulation, including perirenal and epididymal fat. PPAR-γ acts as a major regulator of adipocyte differentiation and metabolism [25,26]. In spontaneously type 2 diabetic rats, telmisartan downsized adipocytes through PPAR-γ-mediated action [9]. Next, GW9662 offset the protective effects of telmisartan on the improvements of glucose metabolism and insulin sensitivity. IPGTT and HOMA-IR also demonstrated that telmisartan exerted a more significant effect on glucose or insulin in HFD mice at 8 weeks than at 4 weeks. Additionally, the protective effect of telmisartan resulted from influences on islet cell mass and apoptosis via activation of the PPAR-γ pathway. Telmisartan could partly abrogate the morphological changes in the pancreatic islets of HFD mice. The alpha/beta ratio and alpha-cell mass were significantly lower in the telmisartan group than in the HFD group. We also showed that telmisartan increased PPAR-γ expression levels in local pancreatic islets. In beta-cells, PPAR-γ regulates a number of the key beta-cell genes, including NKX6.1, Pdx-1, glucokinase and GLUT2 [27,28]. In the adult, Pdx1 contributes to beta-cell gene transcription activation and the maintenance of mature beta-cell functions [29]. Pdx1 also governs the expression of GLUT2 and insulin [29], both of which are markers of beta-cell function. In addition, a recent study shows that telmisartan improves pancreatic ultrastructural enhancement due to the activation of PPAR-γ [12]. Confusingly, we found insulin content markedly decreased while mean islet size and beta-cell mass increased in HFD group. However, it is reasonable that either mean islet size or beta-cell mass in all mice fed high-fat diet (including Telmisartan and GW9662 group) was higher than that in LFD group, which may be due to beta-cell hyperplasia in response to metabolic demand. Last but not least, telmisartan inhibited pancreatic islet inflammation in a PPAR-γ-dependent manner. This inhibition resulted from telmisartan-stimulated PPAR-γ activation, which can modulate macrophage polarization of adipose tissue to an anti-inflammatory M2 state [8]. Interestingly, Kim showed that PPAR-γ activation inhibited NF-kB-dependent cell cytotoxicity to protect beta-cells [30].
In summary, telmisartan, as a partial agonist of PPAR-γ, inhibits insulin resistance, beta-cell dysfunction and islet inflammation and normalizes islet structure in addition to its effects as an ATR1 blocker. These findings suggest that telmisartan therapy could be a practical therapeutic approach for the prevention of insulin resistance and beta-cell dysfunction in Type 2 diabetes.
Acknowledgments
This work was supported by a grant provided by the National Natural Science Foundation of China (no. 81370880).
References
-
Hayden MR, Sowers KM, Pulakat L, Joginpally T, Krueger B, et al. (2011) Possible Mechanisms of Local Tissue Renin-Angiotensin System Activation in the Cardiorenal Metabolic Syndrome and Type 2 Diabetes Mellitus. Cardiorenal Med 1: 193-210.
-
Chappel MC, Ferrario CM (2006) ACE and ACE2: their role to balance the expression of angiotensin II and angiotensin-(1-7). Kidney Int 70: 8-10.
-
Burrell LM, Harrap SB, Velkoska E, Patel SK (2013) The ACE2 gene: its potential as a functional candidate for cardiovascular disease. Clin Sci (Lond) 124: 65-76.
-
Scheen AJ (2004) Renin-angiotensin system inhibition prevents type 2 diabetes mellitus. Part 1. A meta-analysis of randomised clinical trials. Diabetes Metab 30: 487-496.
-
Leung PS (2007) Mechanisms of protective effects induced by blockade of the renin-angiotensin system: novel role of the pancreatic islet angiotensin-generating system in Type 2 diabetes. Diabet Med 24: 110-116.
-
van der Zijl NJ, Moors CC, Goossens GH, Blaak EE, Diamant M (2012) Does interference with the renin-angiotensin system protect against diabetes? Evidence and mechanisms. Diabetes Obes Metab 14: 586-595.
-
Younis F, Oron Y, Limor R, Stern N, Rosenthal T (2012) Prophylactic treatment with telmisartan induces tissue-specific gene modulation favoring normal glucose homeostasis in Cohen-Rosenthal diabetic hypertensive rats. Metabolism 61: 164-174.
-
Fujisaka S, Usui I, Kanatani Y, Ikutani M, Takasaki I, et al. (2011) Telmisartan improves insulin resistance and modulates adipose tissue macrophage polarization in high-fat-fed mice. Endocrinology 152: 1789-1799.
-
Mori Y, Itoh Y, Tajima N (2007) Angiotensin II receptor blockers downsize adipocytes in spontaneously type 2 diabetic rats with visceral fat obesity. Am J Hypertens 20: 431-436.
-
Bhatia V, Viswanathan P (2006) Insulin resistance and PPAR insulin sensitizers. Curr Opin Investig Drugs 7: 891-897.
-
Yamana A, Arita M, Furuta M, Shimajiri Y, Sanke T (2008) The angiotensin II receptor blocker telmisartan improves insulin resistance and has beneficial effects in hypertensive patients with type 2 diabetes and poor glycemic control. Diabetes Res Clin Pract 82: 127-131.
-
Souza-Mello V, Gregorio BM, Relvas-Lucas B, da Silva Faria T, Aguila MB, et al. (2011) Pancreatic ultrastructural enhancement due to telmisartan plus sitagliptin treatment in diet-induced obese C57BL/6 mice. Pancreas 40: 715-722.
-
Lau T, Carlsson PO, Leung PS (2004) Evidence for a local angiotensin-generating system and dose-dependent inhibition of glucose-stimulated insulin release by angiotensin II in isolated pancreatic islets. Diabetologia 47: 240-248.
-
Wang HW, Mizuta M, Saitoh Y, Noma K, Ueno H, et al. (2011) Glucagon-like peptide-1 and candesartan additively improve glucolipotoxicity in pancreatic β-cells. Metabolism 60: 1081-1089.
-
Roat R, Rao V, Doliba NM, Matschinsky FM, Tobias JW, et al. (2014) Alterations of pancreatic islet structure, metabolism and gene expression in diet-induced obese C57BL/6J mice. PLoS One 9: e86815.
-
Fraulob JC, Ogg-Diamantino R, Fernandes-Santos C, Aguila MB, Mandarim-de-Lacerda CA (2010) A Mouse Model of Metabolic Syndrome: Insulin Resistance, Fatty Liver and Non-Alcoholic Fatty Pancreas Disease (NAFPD) in C57BL/6 Mice Fed a High Fat Diet. J Clin Biochem Nutr 46: 212-223.
-
Xiao L, Haack KK, Zucker IH (2013) Angiotensin II regulates ACE and ACE2 in neurons through p38 mitogen-activated protein kinase and extracellular signal-regulated kinase 1/2 signaling. Am J Physiol Cell Physiol 304: C1073-1079.
-
Koka V, Huang XR, Chung AC, Wang W, Truong LD, et al. (2008) Angiotensin II up-regulates angiotensin I-converting enzyme (ACE), but down-regulates ACE2 via the AT1-ERK/p38 MAP kinase pathway. Am J Pathol 172: 1174-1183.
-
Hunyady L, Catt KJ (2006) Pleiotropic AT1 receptor signaling pathways mediating physiological and pathogenic actions of angiotensin II. Mol Endocrinol 20: 953-970.
-
NAVIGATOR Study Group, McMurray JJ, Holman RR, Haffner SM, Bethel MA, et al. (2010) Effect of valsartan on the incidence of diabetes and cardiovascular events. N Engl J Med 362: 1477-1490.
-
Kang YS (2013) Obesity associated hypertension: new insights into mechanism. Electrolyte Blood Press 11: 46-52.
-
Yvan-Charvet L, Quignard-Boulange A (2011) Role of adipose tissue renin-angiotensin system in metabolic and inflammatory diseases associated with obesity. Kidney Int 79: 162-168.
-
Cheng Q, Leung PS (2011) An update on the islet renin-angiotensin system. Peptides 32: 1087-1095.
-
Yuan L, Li Y, Li G, Song Y, Gong X (2013) Ang(1-7) treatment attenuates �-cell dysfunction by improving pancreatic microcirculation in a rat model of Type 2 diabetes. J Endocrinol Invest 36: 931-937.
-
Lefterova MI, Haakonsson AK, Lazar MA, Mandrup S (2014) PPARγ and the global map of adipogenesis and beyond. Trends Endocrinol Metab 25: 293-302.
-
Shih CC, Lin CH, Lin WL (2008) Effects of Momordica charantia on insulin resistance and visceral obesity in mice on high-fat diet. Diabetes Res Clin Pract 81: 134-143.
-
Moibi JA, Gupta D, Jetton TL, Peshavaria M, Desai R, et al. (2007) Peroxisome proliferator-activated receptor-gamma regulates expression of PDX-1 and NKX6.1 in INS-1 cells. Diabetes 56: 88-95.
-
Kim HS, Hwang YC, Koo SH, Park KS, Lee MS, et al. (2013) PPAR- ? activation increases insulin secretion through the up-regulation of the free fatty acid receptor GPR40 in pancreatic β-cells. PLoS One 8: e50128.
-
Arda HE, Benitez CM, Kim SK (2013) Gene regulatory networks governing pancreas development. Dev Cell 25: 5-13.
-
Kim EK, Kwon KB, Koo BS, Han MJ, Song MY, et al. (2007) Activation of peroxisome proliferator-activated receptor-gamma protects pancreatic beta-cells from cytokine-induced cytotoxicity via NF kappaB pathway. Int J Biochem Cell Biol 39: 1260-1275.
