

International Journal of Diabetes and Clinical Research
Novel Insulin Receptor-Signaling Platform
Fiona Haxho1, Farah Alghamdi1,3, Ronald J Neufeld2 and Myron R Szewczuk1,*
1Departments of Biomedical and Molecular Sciences, Queen�s University, Canada
2Chemical Engineering, Queen�s University, Canada
33King Fahd Armed Forces Hospital, Serology, Saudi Arabia
*Corresponding author:
Myron R Szewczuk, Department of Biomedical and Molecular Sciences, Queen's University, Kingston, ON K7L 3N6, Canada, Tel: +1 613 533 2457; Fax: +1 613 533 6796; E-mail: szewczuk@queensu.ca
Int J Diabetes Clin Res, IJDCR-1-005, (Volume 1, Issue 1), Review Article; ISSN: 2377-3634
Received: October 09, 2014 | Accepted: October 27, 2014 | Published: October 30, 2014
Citation: Haxho F, Alghamdi F, Neufeld RJ, Szewczuk MR (2014) Novel Insulin Receptor- Signaling Platform. Int J Diabetes Clin Res 1:005. 10.23937/2377-3634/1410005
Copyright: ©2014 Haxho F, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Insulin receptor (IR) signaling plays a key role in the regulation of glucose homeostasis. A dysfunctional and/or unregulated IR activation has been shown to cause a range of clinical manifestations including insulin resistance, type 2 diabetes, obesity, cancer, hypertension, and cardiovascular disorders. The molecular mechanisms mediating IR activation have become an important area of scientific and clinical research. Here, we summarize the current understanding of IR structure, function, and signaling, and highlight the role of glycosylation and sialylation in IR activation. The key interactions that induce IR activation are identified and a novel IR-signaling platform is proposed. A mammalian neuraminidase-1sialidase (Neu1) and matrix metalloproteinase-9 (MMP-9) cross-talk in alliance with neuromedin B G-protein coupled receptor (GPCR) is uncovered which is essential for insulin-induced IR activation and cellular signaling. Evidence exposing the invisible link connecting insulin-binding to a proposed IR-signaling paradigm will be reviewed in relation to human disease.
Keywords
Insulin receptor, Receptor tyrosine kinase, Glycosylation, neuraminidase-1, Matrix metalloproteinase 9, G-protein-coupled receptor
Introduction
The insulin receptor (IR) is a high affinity transmembrane receptor tyrosine kinase (RTK) complex that is essential in the maintenance of body glucose homeostasis. Although RTK signal transduction is generally well characterized, the framework controlling IR activation remains poorly understood. It is suggested that glycosylation and the regulation of sialylation is required for the control of receptor activation. In support of this, glycosylation of IR is an important modification that allows for the processing, hormonal regulation, and binding activity of the receptor [1]. The specific parameters that mediate IR activation, however, are less clear. Here, we present an overview of insulin receptor structure, function, and the importance of glycosylation in receptor activation and cell signaling. The current understanding of the role of membranebound complexes and receptor modifications associated with IRs will also be reviewed, specifically the role of mammalian neuraminidase-1, matrix metalloproteinase-9, and neuromed in B G-protein coupled receptor. In summary of the research studies examined in this review, we propose a novel insulin receptor-signaling paradigm and discuss its implications in relation to human disease.
Insulin Receptor Structure
The insulin receptor family is a well-characterized group of transmembrane glycoproteins belonging to subclass II of the receptor tyrosine kinase (RTK) superfamily [2]. They consist of the insulin receptor (IR), insulin-like growth factor I receptor (IGFIR), insulin-like growth factor II receptor (IGF-IIR), and IR-related receptor [3]. Insulin-like growth factors (IGFs) are peptide hormones that bind to these receptors and initiate important processes involved in cell growth, development, metabolism and survival. In humans, these peptide hormones comprise of insulin, IGF-I, IGF-II, relaxin, relaxin-like factor (RLF) and placentin [4]. This review will primarily focus on the role of insulin in the activation of IR.
Insulin is a key regulator of metabolic homeostasis, energy storage, and glucose levels [5]. It acts to increase cell glycogen and fatty acid synthesis and esterification, decrease proteolysis, lipolysis and gluconeogenesis, and increase amino acid and potassium uptake [6,7]. Insulin is produced and stored in an inactive form and is released as mature insulin by the pancreatic β-cells via exocytosis directly into hepatic portal circulation. For insulin to bind its receptor, it changes conformation to engage onto its receptor, but the mechanism of which is unclear. Using truncated "microreceptors" that reconstitute the primary hormone-binding site, Menting et al. [8] have shown that insulin�s a-subunit domains L1 and aCT undergo concerted hinge-like rotation at B20-B23 β-turn, thereby coupling reorientation of Phe(B24) to a 60� rotation of the B25-B28 β-strand distant from the insulin core for it to lie antiparallel to the receptor�s L1-β2 sheet. The report also discloses that opening of the insulin hinge enables the conserved nonpolar side chains Ile(A2), Val(A3), Val(B12), Phe(B24), and Phe(B25)) to engage the insulin receptor.
Insulin growth factors-I (IGF-I) and -II are produced by the liver, and are essential for the growth and development of somatic tissues including skeletal muscle and bone [9]. Since IGFs show structural homology to insulin, IGF-IRs are also closely related to IRs [10]. IGF-IR and IR share a common covalently linked 2α2β tetramer structure [11]. The cell surface receptor complex is composed of two extracellular a subunits that contain the insulin binding site, and two transmembrane β subunits that include a cytoplasmic tyrosine kinase (TK) domain [6]. IR and IGF-IR have structurally similar ligand binding domains as well as TK domains [12]. This homologymay provide an explanation for the cross-reaction and functional overlap of these receptors [13]. Notably, IRs binds IGF-I with an approximately 100-fold lower affinity than insulin. Thus, only high concentrations of IGFs can trigger signaling via insulin receptor [14]. A recent report using crystal structure of the related IR dimer with insulin bound has revealed that the ecto domain of the IGF1R maintains an auto-inhibited state in which the transmembrane domains (TMs) are held apart [15]. The data in the report indicated that insulin binding releases this constraint, allowing the association of TMs to facilitate the autophosphorylation of the intracellular regions. Another current report has also shown that IR can be activated through its transmembrane domain [16]. It was shown that a peptidomimetic having sequence identity specific to the IR TM domain can activate IR, but not IGF receptors. The premise behind this IR activation process requires a competent ATP-binding site and kinase activation loop. More interesting, the report also showed results indicating that the IR-TM peptidomimetic can also activate insulin receptors from patients with severe insulin resistance, which do not respond to insulin. Recent advances in azapeptides which are a class of peptidomimetics directed to IR tyrosine kinase domains reveal that in vitro phosphorylation of the these kinases in the presence of Ac-DIazaYET-NH2 was inhibitory whereas the aza-DOPA(3) and aza-Glu(4)peptidomimetics induced IR phosphorylations [17].
IR biosynthesis is a well-characterized process initiated by the synthesis of a single-chain pre-proreceptor, the first product of the insulin receptor gene, INSR [2,18]. Cleavage of the pre-proreceptor forms a proreceptor, which is composed of a covalently linked αβ heterodimer. Subsequently, two αβ complexes will dimerize to form the 2a2β insulin receptor tetramer, which is then localized to the cell surface [6]. IR and IGF-IR have the ability to dimerize and form a heterodimeric IR/IGF-IR complex before migrating to the cell surface4. IRs are translated from a 22 exon sequence from the INSRgene [19]. Depending on the alternative mRNA splicing of exon 11, two different IR isoforms may form [20], which are involved in various metabolic and non-metabolic processes [21]. The IR-A isoform lacks exon 11 and is predominantly expressed during embryogenesis and fetal development, and plays a critical role in promoting embryonic growth and development [22]. In contrast, IR-B includes exon 11 and is predominantly expressed in welldifferentiated tissues, where it plays an important metabolic role in the action of insulin. IR-B up-regulation results in increased signal transduction associated with insulin binding [12].
Various studies indicate that IR-A shows a two-fold higher affinity to bind insulin when compared to IR-B [10,23]. IR-A also has a faster internalization process and recycling time [24]. The higher ligand affinity and faster recycling time associated with IR-A strongly suggests that its upregulation would significantly increase the rate of receptor activation and cell signaling. Despite its higher affinity for insulin when compared to IR-B, the upregulation of IR-A is associated with a much more pronounced increase of insulin growth factor (IGF) binding and signaling [25]. This shift from insulin- to IGF-signaling has been shown to trigger mitogenic and anti-apoptotic signals [10]. Numerous studies have linked IR-A over-expression with human conditions such as diabetes, myotonic dystrophy, and various cancers, including breast cancer, lung cancer and colon cancer [10,12,26,27]. Expectedly, the up-regulation of IR-A in cancer cells has been linked to the over expression of splicing factor hnRNP-A1 and an alteration in the normal balance of IR-A and IR-B [10]. These findings strongly infer a connection between up-regulated IR-signaling and the manifestation of disease. This is supported by other reports that have also implicated the role of these receptors in the development of cancer and diabetes [12,25,27,28]. If this is the case, a disruption of elevated IR-signaling may describe a new target for therapeutic intervention.
Glycosylation is Essential for Insulin Receptor Processing, Localization and Activation
Glycosylation is an important aspect of receptor biosynthesis and functional structure. There are five glycan classes: N-linked glycans, O-linked glycans, phospho-glycans, C-linked glycans, and glycosylphosphatidylinositol (GPI) anchors. Similar to most RTKs, IRs are highly glycosylated receptors; IRs have 18 asparagine residues that function as N-linked glycosylation sites, and some serine and threonine residues involved in O-linked glycosylation [29,30]. Fourteen glycosylation sites are localized on the IRa subunits, while the other sites are on the IRβ subunits [1]. Specifically, the IRa subunits contain only N-linked carbohydrate, while IRβ subunits contain both O- and N-linked carbohydrates [31]. A refined structure of the IR ectodomain that incorporates all of the N-linked glycans and their distribution over the crystal structure has been eloquently reviewed by Sparrow et al. [30].
During IR biosynthesis in the endoplasmic reticulum (ER), N-linked glycans are incorporated into asparagine residues of growing polypeptides [32]. The reversible incorporation of N-linked glycans is controlled by two enzymes: glucosidase and glucosyl transferase [30]. These enzymes are known to play key roles in transient receptor glycosylation at the ER level [33]. The dimerization of IR and the processing of glycans also occur in the ER. Additional glycan modifications occur in the Golgi. Further maturation steps are then required to synthesize a functional IR. These maturation steps include the cleavage of the pre-pro-receptor and the transport of mature IR to the cell surface.
N-linked glycans that are attached to IR have several biological functions. They are essential for polypeptide folding, correct processing of the αβ chain and for trafficking the transportation of mature IR to the cell surface [30]. N-linked glycosylation is also crucial for the oligomerization of complex multimeric proteins [1]. In 2000, Elleman et al. used site-directed mutagenesis to remove N-linked glycosylation sites at 15 different regions of the insulin receptor. Although an individual site mutation did not significantly affect IR function, a combination of mutated glycosylation sites resulted in impaired protein folding and processing [1]. Other reports highlight the importance of N-linked oligosaccharides in the functional structure of insulin receptor [34]. They used an insulin receptor with gene mutations expressed on four N-linked glycosylation sites of the β-subunit (IR-βN1234; ASn-X-Ser/Thr site mutations) and reported a reduction in the molecular weight of IR-βN1234 when compared to the wild-type control. Receptor cell surface expression and insulin binding was not significantly affected. However, IR-βN1234 showed a major disruption of subsequent tyrosine kinase activation and signaling [34].
Cell membrane Sialic Acids and Insulin Resistance
The sialic acid (neuraminic acid) family contains about 50 members that share a common nine-carbon amino sugar backbone. Sialic acids are widely distributed among both invertebrates and vertebrates and are present in all cell surfaces, specifically at the outermost end of glycan chains [35]. All sialic acids are derived from two primary members: 2-keto-5-acetamido-3,5-dideoxy-Dglycero- D-galactononulosonic acid (Neu5Ac) and 2-keto-3-deoxy- D-glycero-D-galactonononicacid (KDN). Among vertebrates, Neu5Ac is the most common sialic acid [36]. Synthesis of sialic acid is initiated in the cytosol and is later transferred to the nucleus and Golgi for further modifications. Sialyl transfetases bind to sialic acid precursors and catalyze the incorporation of specific glycoside linkages. In humans, most sialic acids are linked to galactose as a terminal non-reducing end of glycolipids or glycoproteins [37]. Human insulin receptor contains two types of oligosaccharides: highmannose oligosaccharide and complex oligosaccharide composed of N-acetylglucosamine, fructose, and additional sialic acid residues [1].
The C5 position of sialic acids is considered to be a modification site where distinct functional groups can be attached. At this position, modification processes include acylation, methylation and hydroxylation [38,39]. The modification of C5 is essential for the diversity of sialic acids. This diversity allows sialic acids to perform important biological functions such as cell recognition and contact, neuronal transmission, transportation and localization of proteins, and stabilization of the cell membrane [40]. As negatively charged molecules (due to C1 and C5 functional groups), sialic acids are also able to regulate the transport of positively charged molecules, such as calcium ions [37,41,42]. Furthermore, sialic acids function as recognition sites for host and pathogen receptors which help the immune system to differentiate between self and non-self cells [40]. Their size, negative charge, and exposed terminal position in the carbohydrate chain also allow sialic acids to function as protective shields and to prevent protease degradation of glycoproteins [40].
Studies have demonstrated a connection between insulin resistance and cell membrane sialic acid content. Cytidine 5�-monophospho-N-acetylneuraminic acid (CMP-NANA; substrate used for sialyltransferase sialylation of glycans) was reported to enhance insulin responsiveness by 39%. These findings provide support for the role of cell surface sialic acid in hepatic insulin action with a consequent contribution to insulin resistance associated with diabetes [43]. Other studies implicated the role of sialic acids and the alteration of glycosylation in the development of cancer. Sialic acids have the ability to disguise and conceal recognition sites allowing tumor cells to escape immune detection [36]. One study show that malignant metastatic potential and invasiveness are accompanied by the alteration of sialylation [44]. Other reports indicate that several tumors are sialylated significantly more than normal tissue [40]. In support of this, the increased expression of sialic acids in glycosylated cell surface receptors has been linked to various clinical conditions including malignancies, inflammations, cardiovascular diseases, type I and II diabetes, and insulin resistance [35]. The apparent role of sialylation in receptor signaling and its association to disease may indicate a new molecular target for clinical research.
For tumor growth and survival, glucose contributes an essential nutrient source that supports glycolysis and the hexosamine biosynthesis pathway (HBP). Jones et al. [45] recently reported that insulin-stimulated phosphatidylinositol 3-kinase/protein kinase B (PI3K/PKB) cell survival pathway was strongly abrogated in the absence of extracellular glucose. As a consequence of the loss of insulin-stimulated PI3K activation, short-term glucose deprivation subsequently inhibited tumour cell growth. Loss of insulin-stimulated PKB signalling and cell growth was rescued by extracellular glucosamine and increased activity through the HBP. In addition, they have shown that disruption of O-GlcNAc transferase activity, a terminal step in the HBP, revealed an O-GlcNAcylation process in the PKB signaling and cell growth. Changes in glycosylation are considered a hallmark of cancer, and one of the key targets of glycosylation modifications is E-cadherin. A recent report suggested that insulin and IGF-1 stimulation of breast cancer cells overexpressing E-cadherin induced a decrease of bisecting GlcNAc N-glycans that was accompanied with alterations on E-cadherin cellular localization [46]. These findings provide new evidence for the role insulin/IGF-I signaling pathways contributing to cancer progression through the modification of bisecting GlcNAc N-glycans.
Mammalian Neuraminidases and IR activation
Neuraminidases (sialidases) catalyze the cleavage of sialic acid residues from glycosylated molecules. These enzymes are found in viruses, bacteria, protozoa and mammals. They play key roles in the activation of different receptors, including IRs, IGFRs, and epidermal growth factor receptors (EGFR) [47]. Neuraminidases catalyze the hydrolysis of the terminal sialic acid from oligosaccharides, glycoproteins, and glycolipids, and are capable of cleaving the a-2,3, a-2,4, a-2,6, a-2,8, and a-2,9linkages [48]. The a-2,3 and a-2,6 linkages of sialylated receptors are the most common action sites for hydrolysis [39]. The activity of sialidases has been shown to have critical regulatory functions in the cell, including the ability to mediate proliferation, differentiation, antigenic masking, catabolism, infection and signal transduction [49,50].
Mammalian neuraminidases can be classified into four types (Neu1, Neu2, Neu3 and Neu4)according to their differential localization, optimal pH, response to ions and detergent, kinetic properties and specificity of substrate [37]. Mammalian neuramnidase-1 (Neu1) generally shows the strongest expression in human tissues [51]. Neu1 is located in lysosomes and surface membranes of cells [52,53] and is expressed by the majority of vertebrate tissues, including the pancreas, kidney, heart, lung, liver, brain cells, and skeletal muscle [54]. Neu1 has the ability to form a multi-enzymatic complex that contains two hydrolases: the protective protein cathepsin A (PPCA) and the glycosidase β-galactosidase (β-GAL) [37]. Neu1is the only sialidase known to have a direct involvement in two neurodegenerative metabolic disorders: sialidosis and galactosialidosis [37]. Sialidosis is an inherited metabolic disorder characterized by a clinical deficiency in Neu1 sialidase [55]. Galactosialidosis is caused by the deficiency of both Neu1 and β-GAL [54]. In addition to low levels of Neu1, both disorders involve a metabolic defect that results in the accumulation of sialylation in oligosaccharides and glycoproteins expressed in fibroblast tissues [56]. The involvement of neuraminidase in these conditions calls for a better understanding of its regulatory mechanisms within the cell.
The importance of glycosylation in IR biosynthesis, insulin binding and activation has been studied in detail using site-directed mutagenesis analyses [1,34]. These findings provided supporting evidence for a critical role of oligosaccharide side chains of the IRβ subunit in the molecular events responsible for IR activation and signal transduction. It is well known that membrane glycosylation plays a key role in the function of insulin receptors and glucose transporters. One report disclosed that pretreatment of isolated rat adipocytes with a neuraminidase resulted in a release of sialic acid and an increase in basal glucose transport [57]. This process suggests that sialic acids play a regulatory role in the transport of glucose.
Recently, it has been shown that desialylation of IR enhances insulin-induced cell responses [58]. The treatment of rat skeletal L6 myoblast cells with either mouse-derived mammalian Neu1 sialidase or Clostridium perfringens neuraminidase resulted in a desialylation of IR. This process coincided with a significant increase of L6 myoblast cell proliferation in response to a low dose of insulin [58]. Furthermore, the inhibition of endogenous Neu1 diminished this proliferative effect in the presence of low insulin concentrations (1 and 10 nM), but enhanced proliferation in the presence of higher insulin concentrations (100 nM). For IGF receptors (IGFR), the opposite effect was observed where desialylation of IGFR resulted in an elimination of the heightened proliferative response of L6 myoblasts to 100nM insulin. To explain the contrast in these observations, it was proposed that Neu1 enhanced the mitogenic response of L6 myoblasts to a low dose of insulin. However, the cytosolic Neu2 sialidase and the cell membrane-bound Neu3 sialidase induced myoblast differentiation, but not proliferation, through the direct modulation of the GM3 ganglioside content in myoblasts [59-61] . In support of this premise, it has been reported that a transient up-regulation of Neu3 in L6 myocytes caused a significant decrease in IR signaling. This process was proposed to be in direct modulation of plasma membrane gangliosides by Neu3 activity and the interaction with the growth factor receptor-bound protein 2 (Grb2) [62]. Another explanation for this dichotomy of insulin-induced mitogenic responses may involve GPCR-receptor tyrosine kinase (RTK) novel signaling platforms. Using human embryonic kidney 293 cells, Alderton et al. showed that the platelet-derived growth factor β receptor (PDGFβR) forms a complex with Myc-tagged endothelial differentiation gene-1, a GPCR whose agonist is sphingosine 1-phosphate, in cells co-transfected with these receptors [63]. PDGF plays an important role in the regulation of cell proliferation. PDGF was shown to stimulate tyrosine phosphorylation of the inhibitory Gia subunit to increase p42/p44 mitogen-activated protein kinase (MAPK) activation. Furthermore, the PDGFβR was found to associate with GPCR kinase 2 and β-arrestin-1, both of which play critical roles in the regulation of GPCR signal complex endocytosis - a requirement for the activation of p42/p44 MAPK. The proposition is that PDGFβR signaling is initiated by GPCR kinase 2/β-arrestin-1 complexes that have been recruited to the PDGFβR via tethering to GPCR. These results indicate a novel signaling network characterized by the integration of these receptors that may account for the comitogenic and cell proliferative effects of certain GPCR agonists with PDGF. Notably, basal levels of tyrosine phosphorylated Gia subunits do not induce activation of p42/p44 MAPK on their own [63]. The potentiating effects of Gia signaling on the PDGF-stimulated p42/ p44 MAPK activation may require the PDGF-induced recruitment of other intermediates to tyrosine-phosphorylated Gia subunits. Indeed, several reports have shown that insulin receptors can interact with Gia subunit [64-66]. An alliance between GPCR and RTK is eloquently reviewed by Pyne and colleagues [67-70] and Abdulkhalek et al. [71]. These reviews describe the formation of unique signaling platforms in which protein components specific for each receptor are shared to produce a response upon stimulation with ligand.
The cell membrane-bound Neu3 sialidase has also been shown to be induced by certain standard clinical drugs [72]. Olanzapineis an atypical antipsychotic that belongs to thienobenzodiazepine class of drugs, approved by the U.S. Food and Drug Administration (FDA) for the treatment of patients with schizophrenia and bipolar disorder. Unfortunately, the antipsychotic agent is known to be associated with insulin resistance [73-75]. A recent study reported by us that olanzapine induced Neu3 activity in Neu1-deficient human WG544 or 1140F01 sialidosis fibroblast cells and in Neu4-knockout primary murine macrophages [72]. Olanzapine-induced Neu3 activity also reduced IRβ and IRS1 phosphorylation associated with insulin-stimulated human fibroblast cells. These findings support the premise that a down-regulation of Neu1 by Neu3 activity may lead to impaired IR-signaling associated with insulin-resistance and diabetes [72]. Indeed, Neu3 has been shown to participate in the control of insulin signaling, most likely via the modulation of gangliosides and the interaction with growth factor receptor-bound protein 2 (Grb2) [62]. That report also showed that the transient transfection of Neu3 into 3T3-L1 adipocytes and L6 myocytes caused a significant decrease in IR signaling. Another report has shown that overexpression of Neu3 inhibits MMP-9 expression in vascular smooth muscle cells [76]. When chow-fed mice were given acute and chronic intravenous injections of elastin-binding peptides, they developed hyperglycemia and insulin resistance [77]. The report proposed that this insulin resistance is due to the interaction of IR with Neu1 of the elastin receptor complex triggered by elastin-binding peptides. In another report, it was shown that following elastin peptide treatment, the cellular GM3 levels decreased while lactosylceramide (LacCer) content increased consistently with a GM3/LacCer conversion [78]. It was suggested that Neu1-dependent GM3/LacCer conversion is the key event leading to signaling by the elastin receptor complex. Based on these results, the role of elastin-binding peptides in insulin resistance may involve the direct modulation of plasma membrane gangliosides by increasing Neu3 activity. As described elsewhere, this interaction may be facilitated by the growth factor receptor-bound protein 2 (Grb2) [62]. Recent findings [79] proposing metabolic disorders, such as type 2 diabetes, are membrane microdomain disorders which are caused by aberrant expression of gangliosides fits well with the current findings with GM3 and insulin resistance.
In support of the role of Neu1 in IR activation, a recent report has shown that insulin binding to IR rapidly induced an interaction between IR and Neu1. It was shown that Neu1activity hydrolyzes sialic acid residues of IR and, consequently, induced IR activation [80]. The report also disclosed that when Neu1-deficient mice (expressing ~10% of normal Neu1 activity) were exposed to a high-fat diet, they developed hyperglycemia and insulin resistance compared to the wild-type cohort. The report also proposed that endogenous Neu1 sialidase activity is involved in IR glycosylation modification. Indeed, Blaise et al. provided additional evidence to support that Neu1 interacts with IRβ to desialylate the receptor [77]. Other reports have also suggested that modification of receptor glycosylation may in fact be the connecting link between ligand-binding, receptor dimerization and activation for several other receptors [81-85].
G-protein-Coupled Receptors that Bind Long-Chain Free Fatty Acids and Glucose-Dependent Insulin Secretion
G-protein-coupled receptor 40 (GPR40) is well known as free fatty acid receptor-1. GPR40 is mainly expressed in pancreatic β-cells and it binds medium- and long-chain fatty acids. The activation of GPR40 in these cells causes insulin secretion. The mechanism(s) of receptor activation, pharmacology, and the physiological functions of the fatty acid binding receptors is extensively reviewed by Swaminath [86]. Feng et al. [87] and Talukdar et al. [88] have also reviewed the therapeutic target potential of GPR40in mediating insulin secretion and type 2 diabetes. For an example, a selective GPR40 agonist, TAK-875which is an ago-allosteric modulator of human GPR40, was recently shown to improve glycaemic control by increasing glucose-dependent insulin secretion [89]. The data in the report have also shown that combining metformin which is a first-line drug for treatment of type 2 diabetes and TAK-875 enhanced the glycaemic control in Zucker diabetic fatty rats. This improvement in glycaemic control in this combination group was shown to accompany by a significant increase in fasting plasma insulin levels while the pancreatic insulin content was monitored at levels comparable to those in normal rats (e.g., vehicle: 26, combination: 67.1; normal lean: 69.1 ng�mg-1 pancreas) without affecting pancreatic glucagon content. Using crystal structure analyses of human GPR40 with bound TAK- 875 at 2.3� resolution, the data showed a unique non-canonical binding pocket of TAK-875, which most probably occurs through the lipid bilayer [90]. This crystallized TAK-875-bound GPR40 complex exhibited an inactive-like state. The atomic analyses of the extensive charge network in this ligand binding pocket of GPR40 revealed additional interactions not identified, and thus may provide insights into the plausible binding of multiple ligands to the receptor.
Changes in the metabolic state such as obesity, fasting, cold challenge and high-fat diets (HFDs) are well known to activate complex immune responses. In rodents, HFDs induce a rapid systemic inflammatory response which may lead to obesity. A recent report provided evidence to demonstrate that feeding of high-fat diets to rodents resulted in the increased expression of advanced glycation end products (RAGE) ligand high mobility group box 1 (HMGB1) and carboxymethyllysine-advanced glycation end product epitopes in the liver and adipose tissue [91]. Others have shown that cellular responses to HMGB1 stimulation acting synergistic with oligodeoxynucleotide (ODN) mediated a MyD88-dependent up-regulation of MMP2, MMP9 and cyclin-dependentkinase-2 (CDK2), which was critically dependent on the 35kDa RAGE, the receptor for advanced glycation endproducts and TLR4 receptor [92]. Secreted HMGB1 can be a trigger of inflammation dependent on the complexes it forms with other molecules [93]. Pure recombinant HMGB1 has no proinflammatory activity but can form highly inflammatory complexes with ssDNA, lipopolysaccharide (LPS), IL-1, and nucleosomes, which interact with TLR9, TLR4, IL-1R, and TLR2 receptors, respectively [93]. Interestingly, Ivanov et al. have identified HMGB1 as an ODN� binding protein, and it interacts and pre-associates with TLR9 in the endoplasmic reticulum-Golgi intermediate compartment, hastening TLR9�s redistribution to early endosomes in response to ODN [94]. Based on these data, the extracellular HMGB1 was found to accelerate the delivery of ODNs to its receptor, leading to a TLR9- dependent augmentation of IL-6, IL-12, and TNFa secretion [94]. Others have observed that cells under hyperlipidemic stress relocated HMGB1 protein from the nucleus to cytoplasm [95]. The report also disclosed that the high level of HMGB1 correlated positively with the up-regulation of the RAGE receptors in the lung tissue from hyperlipidemic animals. During hyperlipidemic stress, the HMGB1- RAGE interaction activated the AKT signaling pathway. The cellular and molecular players that participate in the regulation of obesityinduced inflammation and insulin resistance are reviewed by Lee and Lee [96] with particular attention on the roles of the cellular players in these pathogeneses.
Indeed, the pathogenesis for obesity, insulin resistance and type 2 diabetes is usually referred to as a chronic low-grade metabolic meta-inflammation [97]. The contribution of inflammation to insulin resistance has been extensively reviewed [97,98]. Oh et al. [99] have indicated that omega-3 fatty acids bind to the GPR120 receptor in mediating potent anti-inflammatory and insulin-sensitizing effects. The GPR120 receptors bind long-chain fatty acids such as palmitoleic acid, the omega-3 fatty acids (a-linolenic acid, docosahexaenoic acid and eicosapentaenoic acid) [99,100]. In addition, the taste preference for fatty acids is mediated by GPR40 and GPR120 [101]. Oh et al. [99] have described the mechanism of omega-3 fatty acids in mediating anti-inflammatory effects. They have provided evidence to show that omega-3 docosahexaenoic acid (DHA) abolishes lipopolysaccharide (LPS)-mediated phosphorylation and activation of IκB kinase (IKK) and c-Jun N-terminal kinase (JNK) in macrophages but had no effect in macrophages with GPR120 knockdown. It was shown that DHA activation of GPR120 initiates the recruitment of β-arrestin-2 (β-arr-2) to the cytosol and binds to GPR120. β-arr-2 bound to GPR120 interacts with tumor growth factor β (TGF-β) activated kinase-1 binding protein-1 (TAB1), thereby inhibiting the TAB1 interaction with TGF-β activated kinase 1 (TAK1). This process in turn inhibits the downstream pro-inflammatory pathways via activation of NF-κB and JNK. The mechanisms and effects of the concept of "fat taste" in humans [102], and the potential roles of GPR120 and its agonists in the management of diabetes have been extensively reviewed [102-112].
Crosstalk between Insulin Receptor and G Protein- Coupled Receptor Signaling
Crosstalk between insulin receptor and GPCR signaling is being well recognized for the regulation of multiple physiological functions as well as the pathogenesis of important diseases, including cancer, obesity, metabolic syndrome, hypertension, and type II diabetes mellitus, insulin resistance and hyperinsulinemia [113]. The interactions between GPCRs and the large number of glycosylated receptors including the insulin receptor involved in human diseases are quite diverse. Indeed, one GPCR can interact with more than one G-protein to initiate multifunctional signaling. A number of GPCRs can also amplify a response produced by a separate circumstantial signal in the cell. For an example, it has been shown that the activation of TOLL-like receptors (TLRs) can also regulate GPCR responsiveness by modulating the expression of GPCR kinases (GRKs), arrestins (ARRs), and regulators of G-protein-signaling (RGS) proteins [114,115]. GRKs are known to phosphorylate G-proteins while ARRs bind to GPCRs to inhibit G-protein-dependent signaling. Various RGS proteins that act specifically on different Ga subunits and thus stimulate specific pathways are induced by specific TLRs [114]. Together with GRKs and ARRs, RGSs regulate the duration of signaling downstream of GPCRs. The RGS family of proteins, comprising 30 members, is diverse, ranging in size from 17kDa to 160kDa. They display widely variable and regulated expression patterns [116].
Using co-crystallization and atomic structural analyses, it is noteworthy that the GRK compounds can be clustered into two chemical classes; (A)indazole/dihydropyrimidine-containing compounds that are selective for GRK2, and (B) pyrrolopyrimidinecontaining compounds that selectively inhibit GRK1 and GRK5 [117]. The report disclosed interesting evidence to support the selectivity profiles of the most potent inhibitors representing each of the chemical classes, GSK180736A for GRK2 and GSK2163632A for GRK1 and GRK5. GSK180736Awhich develops as a Rho-associated, coiled-coil-containing protein kinase inhibitor, bound to GRK2 similar to that of paroxetine. The other GSK2163632A which develops as an insulin-like growth factor 1 receptor inhibitor, occupied a novel region of the GRK active site cleft. However, neither compound inhibits GRKs more potently than their initial targets.
GPCRs have long been implicated in the transactivation of receptor tyrosine kinases (RTKs) in the absence of their natural growth factors, such as epidermal growth factor, platelet-derived growth factor, fibroblast growth factor, and nerve growth factor [118]. Delcourt and colleagues provided a comprehensive review to explain the molecular mechanisms involved in this cross-talk among GPCRs and RTKs [119]. For an example, insulin-like growth factor-1 (IGF- 1) and the GPCR pituitary adenylyl cyclase activating polypeptide (PACAP) has been shown to increase the phosphorylation state of a common set of proteins in neurons [120]. Using PACAP type 1 receptor (PAC1R) null mice, the report revealed that IGF-1 transactivated PAC1Rs constitutively associated with IGF-1 receptors, and this activation process is mediated by Src family kinases to induce PAC1R phosphorylation on tyrosine residues. Indeed, other reports have shown that IGF-1 can transactivate other GPCRs such as (a) CXC chemokine receptor type 4 (CXCR4) [121], (b) CC chemokine receptor 5 (CCR5) [122], (c) neuropeptide GPCR PAC1 [120], and (d) sphingosine 1-phosphate receptor 1 (S1P1) [123]. In contrast to the general opinion that the trophic activity of IGF-1 is solely mediated by tyrosine kinase receptor-associated signaling, these reports clearly show that it involves a more complex signaling network dependent on the PAC1 Gs-protein-coupled receptor in neurons. Other examples of GPCR-RTK partners have been shown for platelet-derived growth factor (PDGF) [124-126] and nerve growth factor (NGF) [127,128] transactivation of GPCR sphingosine 1-phosphate receptor 1 (S1P1) [123] and NGF transactivate GPCR lysophosphatidic acid receptor 1 (LPA1) [128]. These findings indicate a different receptor signaling concept in which GPCR activation is essential for growth-factor activation of RTK by way of a mechanism that involves a functional signaling complex between the GPCRs and RTK. This crosstalk mechanism has been uncovered by us that describes an entirely new signaling platform [71]. For epidermal growth factor (EGF) receptors [85], EGF binding to its receptor induces a GPCR-signaling process to activate matrix metalloproteinase-9 (MMP-9) and Neu1 sialidase. The report discloses a complex of neuromedin B GPCR, MMP-9 and Neu1 tethered to EGFR at the ectodomain of the receptor on the cell surface. Activated Neu1 specifically hydrolyzes a-2,3-sialyl residues of the receptors, which facilitates the removal of steric hindrance between receptors and allowing subsequent kinase activation and cellular signaling. An identical signaling paradigm has been reported by us for insulin receptors [72], nerve growth factor (NGF) TrkA receptors [129], cell surface TOLL-like receptor-4 (TLR-4) [53,130-133] and intracellular TLR-7 and -9 [71].
For TLRs, we have also shown that GPCR agonists bombesin, LPA, cholesterol, angiotensin-1 and -2, but not thrombin, induce sialidase activity within a minute, and this activity was blocked by Gai-sensitive pertussis toxin and MMP inhibitors galardin and piperazine [131]. It is noteworthy that the GPCR agonists can induce sialidase activity in the live-cell assay but the GPCR-mediated effects were not observed in TLR-deficient cells such as NIH-3T3 cells. These results suggest that GPCR agonists activate sialidase activity only when Neu1 and a functional GPCR are tethered to a TLR receptor. To confirm this hypothesis, bombesin-like receptor neuromedin B (NMBR) was shown to form a complex with not only TLR4 [130] but also with TLR-7 and -9 [134], EGFR [85] and insulin IRβ receptor [72]. In the report on intracellular TLRs, we have shown that NMBR co-immunoprecipitates with MMP9, and conversely, MMP9 co-IPs with NMBR in macrophage cell lysates from naive and TLR9 ligand stimulated cells [134].
There are three bombesin-related peptides that bind three closely related GPCR receptors in mammals, and these GPCRs are neuromedin B-preferring receptor (NMBR), a GRP-preferring receptor (GRPR), and an orphan receptor called bombesin-receptor subtype-3 (BRS-3) [135]. In a study using NMBR knockout female mice, they developed a partial resistance to diet-induced obesity [136]. The study showed that the disruption of the NMBR signaling pathway did not change body weight or food intake in these female mice fed a normolipid diet, but however, they did develop partial resistance to diet-induced obesity, which was not accounted for by alterations in food intake.
It is of interest to note that several GPCRs, for an example, P2Y14(GPCR105) [137], glucagon-like peptide-1 receptor (GLP1R) [138-142], a Zn2+-activated GPCR (GPR39) [143], and M3 muscarinic receptor and galanin receptor [144,145] can modulate the gastrointestinal motility and the glucose-induced insulin secretion.
A Novel GPCR Organizational IR Signaling Platform
A novel GPCR organizational IR signaling platform has recently been uncovered by us and depicted in the graphical abstract (Figure 1). It describes the intermediate players involved in insulin-induced receptor activation [72]. This IR signaling platform constitutes a Neu1- MMP-9 cross-talk initiated by insulin binding to IR. Here, Neu1 and MMP-9 form a complex with neuromedin B GPCR tethered at the ectodomain of IRβ subunits on the cell surface. This signaling paradigm proposes that insulin binding induces a conformational change of the IR to initiate GPCR Gia-signaling and MMP-9 activation. There is substantial evidence to demonstrate that insulin binding to the receptor involves a conformational change through four important processes: (a) a conformational change at the C-terminal domain [146], (b) an increase in the rate of autophosphorylation [147], (c) a fast bimolecular binding reaction followed by a slower unimolecular step representing a conformational change in the insulin-receptor complex[148], and (d) a basal-state conformation of the activation loop that is more accessible overcoming the intra-sterically inhibitory conformation that encompasses =90% of the ligand-free basal state kinase [149]. Analysis of these structures suggests that an insulin-induced conformational change in the receptor may alter the relative orientation of the tyrosine kinases leading to transautophosphorylation and activation of the receptor [150,151]. Based on mutagenesis studies on the four N-glycosylation sites (IRβ-N1234) of the IRβ subunit of the receptor [34], the intra-sterically inhibitory ligand-free basal state kinase is most likely under the control of a-2,3 sialic acid residues of IRβ. Furthermore, the kinase activation state induced by insulin is regulated by Neu1 activity in hydrolyzing these sialic residues and removing steric hindrance of the activation loop as shown by our data [72].
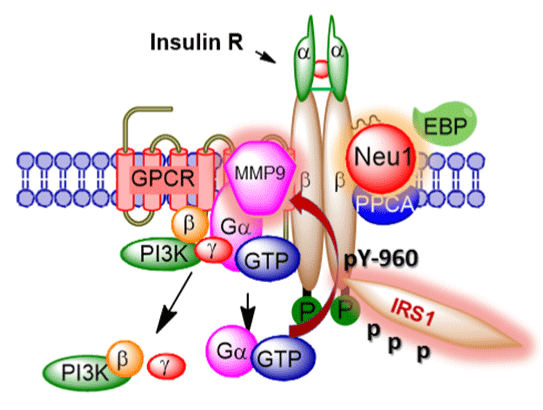
Figure 1: Graphical abstract A novel molecular organizational G-protein-coupled receptor (GPCR)-signaling platform potentiates neuraminidase-1 (Neu1) and matrix metalloproteinase-9
(MMP-9) cross talk on the cell surface that is essential for the activation of
the insulin receptor β subunit (IRβ) tyrosine kinases.
View Figure 1
It can be speculated that insulin binding to IRa on the cell surface initiates GPCR-signaling to activate MMP-9. Our data using coimmunoprecipitation of IRβ and neuromedin B GPCR suggest that the neuromedin B GPCR forms a complex with naive and insulininduced IR in HTC-WT cells [72]. Other co-immunoprecipitation experiments using cell lysates from RAW-Blue cells demonstrated that the 80kDa isoform of the neuromedin B GPCR forms a complex with the active 88 kDa MMP-9 isoform from naive or lipopolysaccharide (LPS)-stimulated cells [131]. These findings validate that neuromedin B GPCR forms a complex with MMP-9 on the cell surface of naive cells. The data in the report also showed that GPCR agonists (bombesin, lysophosphatidic acid, cholesterol, angiotensin-1 and -2, and bradykinin) binding to their respective GPCR receptors induced Neu1 activity within 1 minute and that this Neu1 activity was blocked by Gai-sensitive pertussis toxin, neuraminidase inhibitor oselatmivir phosphate, broad-range MMP inhibitors galardin and piperazine, anti-Neu1 and anti-MMP-9 antibodies, and siRNA knockdown of MMP-9 [131]. The rapidity of the GPCR agonist-induced Neu1 activity suggests that glycosylated receptors such as TLRs and RTKs form a functional GPCR-signaling complex. Indeed, Gilmour et al. [85] and Moody et al. [152] report that the neuromedin B GPCR regulates EGF receptors by a mechanism dependent on MMP activation. It is well known that agonist-bound GPCRs can activate numerous MMPs [153], including MMP-3 [154], MMP-2 and -9 [155,156], and the members of the ADAM family of metalloproteinases [157,158]. We have shown that GPCR agonists can directly activate Neu1 through the intermediate MMP-9 in order to induce transactivation of TLRs and subsequent cellular signaling [131,134]. It is noteworthy that insulin can mediate increases in MMP-9 via IR activation [159], which fits well within our molecular signaling platform of Neu1-MMP-9 cross-talk in regulating insulininduced receptors. That study has also demonstrated that insulin can induce MMP-9 via the mitogenic signaling pathways, whereas the PI3K-dependent signaling which is typically altered in insulin resistance is not required [159]. The connection between GPCR and IR has also been demonstrated for β-adrenergic receptors tethering to IR in adipocytes [160-163]. These reports show that insulin-bound IR stimulates the phosphorylation of the β-adrenergic receptor on Tyr- 350 and this process facilitates IR tethering to β-adrenergic receptor via growth factor receptor-bound protein 2 (Grb-2). This molecular signaling platform integrating the IR/β-adrenergic receptor/Grb-2 tripartite complex is critical for β-adrenergic agonist amplification of insulin-dependent activation of p42/p44 MAPK. The premise behind this molecular signaling platform is to improve IR signaling [160-163], but also to improve signaling of other RTKs associated with GPCR, such as PDGFβR [63]. Reports indicate that PDGF induces a stronger tyrosine phosphorylation of Gai and a more robust activation of p42/ p44 MAPK in cells transfected with both PDGFβR and EDG-1 (the GPCR for SIP-1) compared with PDGFβR alone. These RTK�GPCR signaling platforms are eloquently reviewed by Pyne and Pyne [68].
The GPCR signal integration in IR activation is extensively reviewed by Patel [164] and Abdulkhalek et al. [71] This review presents an overview of the current understanding of insulin receptor structure, the importance of receptor glycosylation and its modifications, and the key intermediate players that are involved in the molecular activation of the insulin IRβ receptor, and subsequent cell signaling. A negative unbalance of this novel IRβ-signaling platform is critical for insulin-induced IR activation and may contribute to insulin resistance and type 2 diabetes [72].
Author's Contributions
F. Haxho and M.R. Szewczuk wrote the paper. M.R. Szewczuk supervised the research design. All authors read and commented on the manuscript.
Author's Information
F. Haxho is the recipient of the Queen's Graduate Award. F. Alghamdi was the recipient of the King Abdullah Scholarship from the Ministry of Higher Education, Saudi Arabia.
References
-
Elleman TC, Frenkel MJ, Hoyne PA, McKern NM, Cosgrove L, et al. (2000) Mutational analysis of the N-linked glycosylation sites of the human insulin receptor. Biochem J 347 Pt 3: 771-779.
-
Renter�a ME, Gandhi NS, Vinuesa P, Helmerhorst E, Mancera RL (2008) A comparative structural bioinformatics analysis of the insulin receptor family ectodomain based on phylogenetic information. PLoS One 3: e3667.
-
Hern�ndez-S�nchez C, Mansilla A, de Pablo F, Zardoya R (2008) Evolution of the insulin receptor family and receptor isoform expression in vertebrates. Mol Biol Evol 25: 1043-1053.
-
De Meyts P, Palsgaard J, Sajid W, Theede AM, Aladdin H (2004) Structural biology of insulin and IGF-1 receptors. Novartis Found Symp 262: 160-171
-
Tatoń J, Czech A, Piatkiewicz P (2010) Insulin as the main regulator of cellular glucose utilization--aetiological aspects of insulin resistance. Endokrynol Pol 61: 388-394.
- Yoshida T, Kawano H, Miyamoto 6. Litwack G (2009) Insulin and IGFs. Elsevier Science.
-
Clark S, Eckardt G, Siddle K, Harrison LC (1991) Changes in insulin-receptor structure associated with trypsin-induced activation of the receptor tyrosine kinase. Biochem J 276 : 27-33.
-
Menting JG, Yang Y, Chan SJ, Phillips NB, Smith BJ, et al. (2014) Protective hinge in insulin opens to enable its receptor engagement. Proc Natl Acad Sci U S A 111: E3395-3404.
-
Maki RG (2010) Small is beautiful: insulin-like growth factors and their role in growth, development, and cancer. J Clin Oncol 28: 4985-4995.
-
Frasca F, Pandini G, Sciacca L, Pezzino V, Squatrito S, et al. (2008) The role of insulin receptors and IGF-I receptors in cancer and other diseases. Arch Physiol Biochem 114: 23-37.
-
Annunziata M, Granata R, Ghigo E (2011) The IGF system. Acta Diabetol 48: 1-9.
-
Belfiore A, Frasca F, Pandini G, Sciacca L, Vigneri R (2009) Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr Rev 30: 586-623.
-
Bertrand L, Horman S, Beauloye C, Vanoverschelde JL (2008) Insulin signalling in the heart. Cardiovasc Res 79: 238-248.
-
Belfiore A, Frasca F (2008) IGF and insulin receptor signaling in breast cancer. J Mammary Gland Biol Neoplasia 13: 381-406.
-
Kavran JM, McCabe JM, Byrne PO, Connacher MK, Wang Z, et al. (2014) How IGF-1 activates its receptor. Elife: 3.
-
Lee J, Miyazaki M, Romeo GR, Shoelson SE (2014) Insulin receptor activation with transmembrane domain ligands. J Biol Chem 289: 19769-19777.
-
Kurian LA, Silva TA, Sabatino D (2014) Submonomer synthesis of azapeptide ligands of the Insulin Receptor Tyrosine Kinase domain. Bioorg Med Chem Lett 24: 4176-4180.
-
Ullrich A, Bell JR, Chen EY, Herrera R, Petruzzelli LM, et al. (1985) Human insulin receptor and its relationship to the tyrosine kinase family of oncogenes. Nature 313: 756-761.
-
Seino S, Seino M, Bell GI (1990) Human insulin-receptor gene. Partial sequence and amplification of exons by polymerase chain reaction. Diabetes 39: 123-128.
-
Zhang H1, Fagan DH, Zeng X, Freeman KT, Sachdev D, et al. (2010) Inhibition of cancer cell proliferation and metastasis by insulin receptor downregulation. Oncogene 29: 2517-2527.
-
Belfiore A, Malaguarnera R (2011) Insulin receptor and cancer. Endocr Relat Cancer 18: R125-147.
-
Chiu SL, Cline HT (2010) Insulin receptor signaling in the development of neuronal structure and function. Neural Dev 5: 7.
-
Lee J, Pilch PF (1994) The insulin receptor: structure, function, and signaling. Am J Physiol 266: C319-334.
-
Lee JK, Tam JW, Tsai MJ, Tsai SY (1992) Identification of cis- and trans-acting factors regulating the expression of the human insulin receptor gene. J Biol Chem 267: 4638-4645.
-
Belfiore A (2007) The role of insulin receptor isoforms and hybrid insulin/IGF-I receptors in human cancer. Curr Pharm Des 13: 671-686.
-
Pollak M, Russell-Jones D (2010) Insulin analogues and cancer risk: cause for concern or cause c�l�bre? Int J Clin Pract 64: 628-636.
-
Mussig K, Staiger H, Kantartzis K, Fritsche A, Kanz L, et al. (2011) Type 2 diabetes mellitus and risk of malignancy: is there a strategy to identify a subphenotype of patients with increased susceptibility to endogenous and exogenous hyperinsulinism? Diabet Med 28: 276-286.
-
Azar M, Lyons TJ (2010) Diabetes, insulin treatment, and cancer risk: what is the evidence? F1000 Med Rep 2.
-
Ward C, Lawrence M, Streltsov V, Garrett T, McKern N, et al. (2008) Structural insights into ligand-induced activation of the insulin receptor. Acta Physiol (Oxf) 192: 3-9.
-
Sparrow LG, Lawrence MC, Gorman JJ, Strike PM, Robinson CP, et al. (2008) N-linked glycans of the human insulin receptor and their distribution over the crystal structure. Proteins 71: 426-439.
-
Mayer JP, Zhang F, DiMarchi RD (2007) Insulin structure and function. Biopolymers 88: 687-713.
-
Fernandez F, Jannatipour M, Hellman U, Rokeach LA, Parodi AJ (1996) A new stress protein: synthesis of Schizosaccharomyces pombe UDP--Glc:glycoprotein glucosyltransferase mRNA is induced by stress conditions but the enzyme is not essential for cell viability. EMBO J 15: 705-713
-
Kornfeld R, Kornfeld S (1985) Assembly of asparagine-linked oligosaccharides. Annu Rev Biochem 54: 631-664.
-
Leconte I, Auzan C, Debant A, Rossi B, Clauser E (1992) N-linked oligosaccharide chains of the insulin receptor beta subunit are essential for transmembrane signaling. J Biol Chem 267: 17415-17423.
-
Varki A (2008) Sialic acids in human health and disease. Trends Mol Med 14: 351-360.
-
Schwerdtfeger SM, Melzig MF (2010) Sialidases in biological systems. Pharmazie 65: 551-561.
-
Monti E, Bonten E, D'Azzo A, Bresciani R, Venerando B, et al. (2010) Sialidases in vertebrates: a family of enzymes tailored for several cell functions. Adv Carbohydr Chem Biochem 64: 403-479.
-
Buschiazzo A, Alzari PM (2008) Structural insights into sialic acid enzymology. Curr Opin Chem Biol 12: 565-572.
-
Achyuthan KE, Achyuthan AM (2001) Comparative enzymology, biochemistry and pathophysiology of human exo-alpha-sialidases (neuraminidases). Comp Biochem Physiol B Biochem Mol Biol 129: 29-64.
-
Traving C, Schauer R (1998) Structure, function and metabolism of sialic acids. Cell Mol Life Sci 54: 1330-1349.
-
Schauer R (2000) Achievements and challenges of sialic acid research. Glycoconj J 17: 485-499.
-
Nakano V, Fontes Piazza RM, Avila-Campos MJ (2006) A rapid assay of the sialidase activity in species of the Bacteroides fragilis group by using peanut lectin hemagglutination. Anaerobe 12: 238-241.
-
Salhanick AI, Amatruda JM (1988) Role of sialic acid in insulin action and the insulin resistance of diabetes mellitus. Am J Physiol 255: E173-179.
-
Miyagi T, Wada T, Yamaguchi K, Shiozaki K, Sato I, et al. (2008) Human sialidase as a cancer marker. Proteomics 8: 3303-3311.
-
Jones DR, Keune WJ, Anderson KE, Stephens LR, Hawkins PT, et al. (2014) The hexosamine biosynthesis pathway and O-GlcNAcylation maintain insulin-stimulated PI3K-PKB phosphorylation and tumour cell growth after short-term glucose deprivation. FEBS J 281: 3591-3608.
-
de-Freitas-Junior JC, Carvalho S, Dias AM, Oliveira P, Cabral J, et al. (2013) Insulin/IGF-I signaling pathways enhances tumor cell invasion through bisecting GlcNAc N-glycans modulation. an interplay with E-cadherin. PLoS One 8: e81579.
-
Hinek A, Bodnaruk TD, Bunda S, Wang Y, Liu K (2008) Neuraminidase-1, a subunit of the cell surface elastin receptor, desialylates and functionally inactivates adjacent receptors interacting with the mitogenic growth factors PDGF-BB and IGF-2. Am J Pathol 173: 1042-1056.
-
Magesh S, Moriya S, Suzuki T, Miyagi T, Ishida H, et al. (2008) Design, synthesis, and biological evaluation of human sialidase inhibitors. Part 1: selective inhibitors of lysosomal sialidase (NEU1). Bioorg Med Chem Lett 18: 532-537.
-
Magesh S, Suzuki T, Miyagi T, Ishida H, Kiso M (2006) Homology modeling of human sialidase enzymes NEU1, NEU3 and NEU4 based on the crystal structure of NEU2: hints for the design of selective NEU3 inhibitors. J Mol Graph Model 25: 196-207.
-
Monti E, Preti A, Venerando B, Borsani G (2002) Recent development in mammalian sialidase molecular biology. Neurochem Res 27: 649-663.
-
Hata K, Koseki K, Yamaguchi K, Moriya S, Suzuki Y, et al. (2008) Limited inhibitory effects of oseltamivir and zanamivir on human sialidases. Antimicrob Agents Chemother 52: 3484-3491.
-
Pshezhetsky AV, Hinek A (2011) Where catabolism meets signalling: neuraminidase 1 as a modulator of cell receptors. Glycoconj J 28: 441-452.
-
Amith SR, Jayanth P, Franchuk S, Finlay T, Seyrantepe V, et al. (2010) Neu1 desialylation of sialyl alpha-2,3-linked beta-galactosyl residues of TOLL-like receptor 4 is essential for receptor activation and cellular signaling. Cell Signal 22: 314-324.
-
Bonten E, van der Spoel A, Fornerod M, Grosveld G, d'Azzo A (1996) Characterization of human lysosomal neuraminidase defines the molecular basis of the metabolic storage disorder sialidosis. Genes Dev 10: 3156-3169.
-
Seyrantepe V, Poupetova H, Froissart R, Zabot MT, Maire I, et al. (2003) Molecular pathology of NEU1 gene in sialidosis. Hum Mutat 22: 343-352.
-
van Pelt J, Kamerling JP, Vliegenthart JF, Hoogeveen AT, Galjaard H (1988) A comparative study of the accumulated sialic acid-containing oligosaccharides from cultured human galactosialidosis and sialidosis fibroblasts. Clin Chim Acta 174: 325-335.
-
Ciaraldi TP (1989) Neuraminidase treatment of isolated rat adipocytes and differential regulation of basal and insulin-stimulated glucose transport. Diabetes 38: 951-958.
-
Arabkhari M, Bunda S, Wang Y, Wang A, Pshezhetsky AV, et al. (2010) Desialylation of insulin receptors and IGF-1 receptors by neuraminidase-1 controls the net proliferative response of L6 myoblasts to insulin. Glycobiology 20: 603-616.
-
Fanzani A, Giuliani R, Colombo F, Zizioli D, Presta M, et al. (2003) Overexpression of cytosolic sialidase Neu2 induces myoblast differentiation in C2C12 cells. FEBS Lett 547: 183-188.
-
Anastasia L, Papini N, Colazzo F, Palazzolo G, Tringali C, et al. (2008) NEU3 sialidase strictly modulates GM3 levels in skeletal myoblasts C2C12 thus favoring their differentiation and protecting them from apoptosis. J Biol Chem 283: 36265-36271.
-
Sato K, Miyagi T (1996) Involvement of an endogenous sialidase in skeletal muscle cell differentiation. Biochem Biophys Res Commun 221: 826-830.
-
Sasaki A, Hata K, Suzuki S, Sawada M, Wada T, et al. (2003) Overexpression of plasma membrane-associated sialidase attenuates insulin signaling in transgenic mice. J Biol Chem 278: 27896-27902.
-
Alderton F, Rakhit S, Kong KC, Palmer T, Sambi B, et al. (2001) Tethering of the platelet-derived growth factor beta receptor to G-protein-coupled receptors. A novel platform for integrative signaling by these receptor classes in mammalian cells. J Biol Chem 276: 28578-28585.
-
Rothenberg PL, Kahn CR (1988) Insulin inhibits pertussis toxin-catalyzed ADP-ribosylation of G-proteins. Evidence for a novel interaction between insulin receptors and G-proteins. J Biol Chem 263: 15546-15552.
-
Luttrell L, Kilgour E, Larner J, Romero G (1990) A pertussis toxin-sensitive G-protein mediates some aspects of insulin action in BC3H-1 murine myocytes. J Biol Chem 265: 16873-16879.
-
O'Brien RM, Houslay MD, Milligan G, Siddle K (1987) The insulin receptor tyrosyl kinase phosphorylates holomeric forms of the guanine nucleotide regulatory proteins Gi and Go. FEBS Lett 212: 281-288.
-
Pyne NJ, Pyne S (2008) Sphingosine 1-phosphate, lysophosphatidic acid and growth factor signaling and termination. Biochim Biophys Acta 1781: 467-476.
-
Pyne NJ, Pyne S (2011) Receptor tyrosine kinase-G-protein-coupled receptor signalling platforms: out of the shadow? Trends Pharmacol Sci 32: 443-450.
-
Pyne NJ, Waters C, Moughal NA, Sambi BS, Pyne S (2003) Receptor tyrosine kinase-GPCR signal complexes. Biochem Soc Trans 31: 1220-1225.
-
Pyne NJ, Waters CM, Long JS, Moughal NA, Tigyi G, et al. (2007) Receptor tyrosine kinase-G-protein coupled receptor complex signaling in mammalian cells. Adv Enzyme Regul 47: 271-280.
-
Abdulkhalek S, Hrynyk, M., Szewczuk, M.R (2013) A novel G-protein-coupled receptor-signaling platform and its targeted translation in human disease. Research and Reports in Biochemistry 3: 17-30.
-
Alghamdi F, Guo M, Abdulkhalek S, Crawford N, Amith SR, et al. (2014) A novel insulin receptor-signaling platform and its link to insulin resistance and type 2 diabetes. Cell Signal 26: 1355-1368.
-
Ebenbichler CF, Laimer M, Eder U, Mangweth B, Weiss E, et al. (2003) Olanzapine induces insulin resistance: results from a prospective study. J Clin Psychiatry 64: 1436-1439.
-
Richards AA, Hickman IJ, Wang AY, Jones AL, Newell F, et al. (2006) Olanzapine treatment is associated with reduced high molecular weight adiponectin in serum: a potential mechanism for olanzapine-induced insulin resistance in patients with schizophrenia. J Clin Psychopharmacol 26: 232-237.
-
Engl J, Laimer M, Niederwanger A, Kranebitter M, Starzinger M, et al. (2005) Olanzapine impairs glycogen synthesis and insulin signaling in L6 skeletal muscle cells. Mol Psychiatry 10: 1089-1096.
-
Moon SK, Cho SH, Kim KW, Jeon JH, Ko JH, et al. (2007) Overexpression of membrane sialic acid-specific sialidase Neu3 inhibits matrix metalloproteinase-9 expression in vascular smooth muscle cells. Biochem Biophys Res Commun 356: 542-547.
-
Blaise S, Romier B, Kawecki C, Ghirardi M, Rabenoelina F, et al. (2013) Elastin-derived peptides are new regulators of insulin resistance development in mice. Diabetes 62: 3807-3816.
-
Rusciani A, Duca L, Sartelet H, Chatron-Colliet A, Bobichon H, et al. (2010) Elastin peptides signaling relies on neuraminidase-1-dependent lactosylceramide generation. PLoS One 5: e14010.
-
Inokuchi J (2010) Membrane microdomains and insulin resistance. FEBS Lett 584: 1864-1871.
-
Dridi L, Seyrantepe V, Fougerat A, Pan X, Bonneil E, et al. (2013) Positive regulation of insulin signaling by neuraminidase 1. Diabetes 62: 2338-2346.
-
Slieker LJ, Lane MD (1985) Post-translational processing of the epidermal growth factor receptor. Glycosylation-dependent acquisition of ligand-binding capacity. J Biol Chem 260: 687-690.
-
Soderquist AM, Carpenter G (1984) Glycosylation of the epidermal growth factor receptor in A-431 cells. The contribution of carbohydrate to receptor function. J Biol Chem 259: 12586-12594.
-
Takahashi M, Tsuda T, Ikeda Y, Honke K, Taniguchi N (2004) Role of N-glycans in growth factor signaling. Glycoconj J 20: 207-212.
-
Fernandes H, Cohen S, Bishayee S (2001) Glycosylation-induced conformational modification positively regulates receptor-receptor association: a study with an aberrant epidermal growth factor receptor (EGFRvIII/DeltaEGFR) expressed in cancer cells. J Biol Chem 276: 5375-5383.
-
Gilmour AM, Abdulkhalek S, Cheng TS, Alghamdi F, Jayanth P, et al. (2013) A novel epidermal growth factor receptor-signaling platform and its targeted translation in pancreatic cancer. Cell Signal 25: 2587-2603.
-
Swaminath G (2008) Fatty acid binding receptors and their physiological role in type 2 diabetes. Arch Pharm (Weinheim) 341: 753-761.
-
Feng XT, Leng J, Xie Z, Li SL, Zhao W, et al. (2012) GPR40: a therapeutic target for mediating insulin secretion (review). Int J Mol Med 30: 1261-1266.
-
Talukdar S, Olefsky JM, Osborn O (2011) Targeting GPR120 and other fatty acid-sensing GPCRs ameliorates insulin resistance and inflammatory diseases. Trends Pharmacol Sci 32: 543-550.
-
Ito R, Tsujihata Y, Matsuda-Nagasumi K, Mori I, Negoro N, et al. (2013) TAK-875, a GPR40/FFAR1 agonist, in combination with metformin prevents progression of diabetes and β-cell dysfunction in Zucker diabetic fatty rats. Br J Pharmacol 170: 568-580.
-
Srivastava A, Yano J, Hirozane Y, Kefala G, Gruswitz F, et al. (2014) High-resolution structure of the human GPR40 receptor bound to allosteric agonist TAK-875. Nature 513: 124-127.
-
Song F, Hurtado del Pozo C, Rosario R, Zou YS, Ananthakrishnan R, et al. (2014) RAGE regulates the metabolic and inflammatory response to high-fat feeding in mice. Diabetes 63: 1948-1965.
-
Wang C, Fei G, Liu Z, Li Q, Xu Z, et al. (2012) HMGB1 was a pivotal synergistic effecor for CpG oligonucleotide to enhance the progression of human lung cancer cells. Cancer Biol Ther 13: 727-736.
-
Bianchi ME (2009) HMGB1 loves company. J Leukoc Biol 86: 573-576.
-
Ivanov S, Dragoi AM, Wang X, Dallacosta C, Louten J, et al. (2007) A novel role for HMGB1 in TLR9-mediated inflammatory responses to CpG-DNA. Blood 110: 1970-1981.
-
Haraba R, Suica VI, Uyy E, Ivan L, Antohe F (2011) Hyperlipidemia stimulates the extracellular release of the nuclear high mobility group box 1 protein. Cell Tissue Res 346: 361-368.
-
Lee BC, Lee J (2014) Cellular and molecular players in adipose tissue inflammation in the development of obesity-induced insulin resistance. Biochim Biophys Acta 1842: 446-462.
-
Gregor MF, Hotamisligil GS (2011) Inflammatory mechanisms in obesity. Annu Rev Immunol 29: 415-445.
-
Donath MY, Shoelson SE (2011) Type 2 diabetes as an inflammatory disease. Nat Rev Immunol 11: 98-107.
-
Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, et al. (2010) GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 142: 687-698.
-
Hirasawa A, Tsumaya K, Awaji T, Katsuma S, Adachi T, et al. (2005) Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat Med 11: 90-94.
-
Cartoni C, Yasumatsu K, Ohkuri T, Shigemura N, Yoshida R, et al. (2010) Taste preference for fatty acids is mediated by GPR40 and GPR120. J Neurosci 30: 8376-8382.
-
Tucker RM, Mattes RD, Running CA (2014) Mechanisms and effects of "fat taste" in humans. Biofactors 40: 313-326.
-
Abdoul-Azize S, Selvakumar S, Sadou H, Besnard P, Khan NA (2014) Ca2+ signaling in taste bud cells and spontaneous preference for fat: unresolved roles of CD36 and GPR120. Biochimie 96: 8-13.
-
Yonezawa T, Kurata R, Yoshida K, Murayama MA, Cui X, et al. (2013) Free fatty acids-sensing G protein-coupled receptors in drug targeting and therapeutics. Curr Med Chem 20: 3855-3871.
-
Halder S, Kumar S, Sharma R (2013) The therapeutic potential of GPR120: a patent review. Expert Opin Ther Pat 23: 1581-1590.
-
Gilbertson TA, Khan NA (2014) Cell signaling mechanisms of oro-gustatory detection of dietary fat: advances and challenges. Prog Lipid Res 53: 82-92.
-
Cornall LM, Mathai ML, Hryciw DH, McAinch AJ (2014) GPR120 agonism as a countermeasure against metabolic diseases. Drug Discov Today 19: 670-679.
-
Hara T, Kashihara D, Ichimura A, Kimura I, Tsujimoto G, et al. (2014) Role of free fatty acid receptors in the regulation of energy metabolism. Biochim Biophys Acta 1841: 1292-1300.
-
Thorburn AN, Macia L, Mackay CR (2014) Diet, metabolites, and "western-lifestyle" inflammatory diseases. Immunity 40: 833-842.
-
Ichimura A, Hara T, Hirasawa A (2014) Regulation of Energy Homeostasis via GPR120. Front Endocrinol (Lausanne) 5: 111.
-
Zhang D, Leung PS (2014) Potential roles of GPR120 and its agonists in the management of diabetes. Drug Des Devel Ther 8: 1013-1027.
-
Calder PC (2014) Marine omega-3 fatty acids and inflammatory processes: Effects, mechanisms and clinical relevance. Biochim Biophys Acta .
-
Velloso LA, Folli F, Perego L, Saad MJ (2006) The multi-faceted cross-talk between the insulin and angiotensin II signaling systems. Diabetes Metab Res Rev 22: 98-107.
-
Lattin J, Zidar DA, Schroder K, Kellie S, Hume DA, et al. (2007) G-protein-coupled receptor expression, function, and signaling in macrophages. J Leukoc Biol 82: 16-32.
-
Loniewski K, Shi Y, Pestka J, Parameswaran N (2008) Toll-like receptors differentially regulate GPCR kinases and arrestins in primary macrophages. Mol Immunol 45: 2312-2322.
-
Wieland T, Mittmann C (2003) Regulators of G-protein signalling: multifunctional proteins with impact on signalling in the cardiovascular system. Pharmacol Ther 97: 95-115.
-
Homan KT, Larimore KM, Elkins JM, Szklarz M, Knapp S, Tesmer JJ (2014) Identification and Structure-Function Analysis of Subfamily Selective G Protein-Coupled Receptor Kinase Inhibitors. ACS chemical biology.
-
Shah BH, Catt KJ (2004) GPCR-mediated transactivation of RTKs in the CNS: mechanisms and consequences. Trends Neurosci 27: 48-53.
-
Delcourt N, Bockaert J, Marin P (2007) GPCR-jacking: from a new route in RTK signalling to a new concept in GPCR activation. Trends Pharmacol Sci 28: 602-607.
-
Delcourt N, Thouvenot E, Chanrion B, Gal�otti N, Jouin P, et al. (2007) PACAP type I receptor transactivation is essential for IGF-1 receptor signalling and antiapoptotic activity in neurons. EMBO J 26: 1542-1551.
-
Akekawatchai C, Holland JD, Kochetkova M, Wallace JC, McColl SR (2005) Transactivation of CXCR4 by the insulin-like growth factor-1 receptor (IGF-1R) in human MDA-MB-231 breast cancer epithelial cells. J Biol Chem 280: 39701-39708.
-
Mira E, Lacalle RA, Gonz�lez MA, G�mez-Mout�n C, Abad JL, et al. (2001) A role for chemokine receptor transactivation in growth factor signaling. EMBO Rep 2: 151-156.
-
El-Shewy HM, Johnson KR, Lee MH, Jaffa AA, Obeid LM, et al. (2006) Insulin-like growth factors mediate heterotrimeric G protein-dependent ERK1/2 activation by transactivating sphingosine 1-phosphate receptors. J Biol Chem 281: 31399-31407.
-
Rosenfeldt HM, Hobson JP, Milstien S, Spiegel S (2001) The sphingosine-1-phosphate receptor EDG-1 is essential for platelet-derived growth factor-induced cell motility. Biochem Soc Trans 29: 836-839.
-
Rosenfeldt HM, Hobson JP, Maceyka M, Olivera A, Nava VE, et al. (2001) EDG-1 links the PDGF receptor to Src and focal adhesion kinase activation leading to lamellipodia formation and cell migration. FASEB J 15: 2649-2659.
-
Hobson JP, Rosenfeldt HM, Barak LS, Olivera A, Poulton S, et al. (2001) Role of the sphingosine-1-phosphate receptor EDG-1 in PDGF-induced cell motility. Science 291: 1800-1803.
-
Toman RE, Payne SG, Watterson KR, Maceyka M, Lee NH, et al. (2004) Differential transactivation of sphingosine-1-phosphate receptors modulates NGF-induced neurite extension. J Cell Biol 166: 381-392.
-
Moughal NA, Waters C, Sambi B, Pyne S, Pyne NJ (2004) Nerve growth factor signaling involves interaction between the Trk A receptor and lysophosphatidate receptor 1 systems: nuclear translocation of the lysophosphatidate receptor 1 and Trk A receptors in pheochromocytoma 12 cells. Cell Signal 16: 127-136.
-
Jayanth P, Amith SR, Gee K, Szewczuk MR (2010) Neu1 sialidase and matrix metalloproteinase-9 cross-talk is essential for neurotrophin activation of Trk receptors and cellular signaling. Cell Signal 22: 1193-1205.
-
Abdulkhalek S, Amith SR, Franchuk SL, Jayanth P, Guo M, et al. (2011) Neu1 sialidase and matrix metalloproteinase-9 cross-talk is essential for Toll-like receptor activation and cellular signaling. J Biol Chem 286: 36532-36549.
-
Abdulkhalek S, Guo M, Amith SR, Jayanth P, Szewczuk MR (2012) G-protein coupled receptor agonists mediate Neu1 sialidase and matrix metalloproteinase-9 cross-talk to induce transactivation of TOLL-like receptors and cellular signaling. Cell Signal 24: 2035-2042.
-
Amith SR, Jayanth P, Finlay T, Franchuk S, Gilmour A, et al. (2010) Detection of Neu1 sialidase activity in regulating Toll-like receptor activation. J Vis Exp
-
Amith SR, Jayanth P, Franchuk S, Siddiqui S, Seyrantepe V, et al. (2009) Dependence of pathogen molecule-induced toll-like receptor activation and cell function on Neu1 sialidase. Glycoconj J 26: 1197-1212.
-
Abdulkhalek S, Szewczuk MR (2013) Neu1 sialidase and matrix metalloproteinase-9 cross-talk regulates nucleic acid-induced endosomal TOLL-like receptor-7 and -9 activation, cellular signaling and pro-inflammatory responses. Cell Signal 25: 2093-2105.
-
Weber HC (2009) Regulation and signaling of human bombesin receptors and their biological effects. Curr Opin Endocrinol Diabetes Obes 16: 66-71.
-
Paula GS, Souza LL, Cabanelas A, Bloise FF, Mello-Coelho V, et al. (2010) Female mice target deleted for the neuromedin B receptor have partial resistance to diet-induced obesity. J Physiol 588: 1635-1645.
-
Meister J, Le Duc D, Ricken A, Burkhardt R, Thiery J, et al. (2014) The G protein-coupled receptor P2Y14 influences insulin release and smooth muscle function in mice. J Biol Chem 289: 23353-23366.
-
Jun LS, Millican RL, Hawkins ED, Konkol DL, Showalter AD, et al. (2014) Absence of Glucagon and Insulin Action Reveals a Role for the Glucagon-like Peptide-1 Receptor in Endogenous Glucose Production. Diabetes .
-
Ross SA (2014) A multiplicity of targets: evaluating composite endpoint studies of the GLP-1 receptor agonists in type 2 diabetes. Curr Med Res Opin .
-
Fern�ndez-Garc�a JC, Colomo N, Tinahones FJ (2014) [Effects of GLP-1 receptor agonists on carbohydrate metabolism control]. Med Clin (Barc) 143 Suppl 2: 18-22.
-
Iepsen EW, Torekov SS, Holst JJ (2014) Therapies for inter-relating diabetes and obesity - GLP-1 and obesity. Expert Opin Pharmacother .
-
Eng C, Kramer CK, Zinman B, Retnakaran R (2014) Glucagon-like peptide-1 receptor agonist and basal insulin combination treatment for the management of type 2 diabetes: a systematic review and meta-analysis. Lancet .
-
Popovics P, Stewart AJ (2011) GPR39: a Zn(2+)-activated G protein-coupled receptor that regulates pancreatic, gastrointestinal and neuronal functions. Cell Mol Life Sci 68: 85-95.
-
Gautam D, Han SJ, Duttaroy A, Mears D, Hamdan FF, et al. (2007) Role of the M3 muscarinic acetylcholine receptor in beta-cell function and glucose homeostasis. Diabetes Obes Metab 9 Suppl 2: 158-169.
-
Duttaroy A, Zimliki CL, Gautam D, Cui Y, Mears D, et al. (2004) Muscarinic stimulation of pancreatic insulin and glucagon release is abolished in m3 muscarinic acetylcholine receptor-deficient mice. Diabetes 53: 1714-1720.
-
Baron V, Gautier N, Komoriya A, Hainaut P, Scimeca JC, et al. (1990) Insulin binding to its receptor induces a conformational change in the receptor C-terminus. Biochemistry 29: 4634-4641.
-
Lee J, Pilch PF, Shoelson SE, Scarlata SF (1997) Conformational changes of the insulin receptor upon insulin binding and activation as monitored by fluorescence spectroscopy. Biochemistry 36: 2701-2708.
-
Schlein M, Havelund S, Kristensen C, Dunn MF, Kaarsholm NC (2000) Ligand-induced conformational change in the minimized insulin receptor. J Mol Biol 303: 161-169.
-
Frankel M, Bishop SM, Ablooglu AJ, Han YP, Kohanski RA (1999) Conformational changes in the activation loop of the insulin receptor's kinase domain. Protein Sci 8: 2158-2165.
-
Hubbard SR, Wei L, Ellis L, Hendrickson WA (1994) Crystal structure of the tyrosine kinase domain of the human insulin receptor. Nature 372: 746-754.
-
McDonald NQ, Murray-Rust J, Blundell TL (1995) The first structure of a receptor tyrosine kinase domain: a further step in understanding the molecular basis of insulin action. Structure 3: 1-6.
-
Moody TW, Berna MJ, Mantey S, Sancho V, Ridnour L, et al. (2010) Neuromedin B receptors regulate EGF receptor tyrosine phosphorylation in lung cancer cells. Eur J Pharmacol 637: 38-45.
-
Fischer OM, Hart S, Ullrich A (2006) Dissecting the epidermal growth factor receptor signal transactivation pathway. Methods Mol Biol 327: 85-97.
-
Lee MH, Murphy G (2004) Matrix metalloproteinases at a glance. J Cell Sci 117: 4015-4016.
-
Le Gall SM, Auger R, Dreux C, Mauduit P (2003) Regulated cell surface pro-EGF ectodomain shedding is a zinc metalloprotease-dependent process. J Biol Chem 278: 45255-45268.
-
Murasawa S, Mori Y, Nozawa Y, Gotoh N, Shibuya M, et al. (1998) Angiotensin II type 1 receptor-induced extracellular signal-regulated protein kinase activation is mediated by Ca2+/calmodulin-dependent transactivation of epidermal growth factor receptor. Circ Res 82: 1338-1348.
-
Gooz M, Gooz P, Luttrell LM, Raymond JR (2006) 5-HT2A Receptor Induces ERK Phosphorylation and Proliferation through ADAM-17 Tumor Necrosis Factor-{alpha}-converting Enzyme (TACE) Activation and Heparin-bound Epidermal Growth Factor-like Growth Factor (HB-EGF) Shedding in Mesangial Cells. J Biol Chem 281: 21004-21012.
-
Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, et al. (1999) EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature 402: 884-888.
-
Fischoeder A, Meyborg H, Stibenz D, Fleck E, Graf K, et al. (2007) Insulin augments matrix metalloproteinase-9 expression in monocytes. Cardiovasc Res 73: 841-848.
-
Karoor V, Wang L, Wang HY, Malbon CC (1998) Insulin stimulates sequestration of beta-adrenergic receptors and enhanced association of beta-adrenergic receptors with Grb2 via tyrosine 350. J Biol Chem 273: 33035-33041.
-
Karoor V, Malbon CC (1996) Insulin-like growth factor receptor-1 stimulates phosphorylation of the beta2-adrenergic receptor in vivo on sites distinct from those phosphorylated in response to insulin. J Biol Chem 271: 29347-29352.
-
Baltensperger K, Karoor V, Paul H, Ruoho A, Czech MP, et al. (1996) The beta-adrenergic receptor is a substrate for the insulin receptor tyrosine kinase. J Biol Chem 271: 1061-1064.
-
Karoor V, Baltensperger K, Paul H, Czech MP, Malbon CC (1995) Phosphorylation of tyrosyl residues 350/354 of the beta-adrenergic receptor is obligatory for counterregulatory effects of insulin. J Biol Chem 270: 25305-25308
-
Patel TB (2004) Single transmembrane spanning heterotrimeric g protein-coupled receptors and their signaling cascades. Pharmacol Rev 56: 371-385.
