

International Journal of Cancer and Clinical Research
Colorectal Cancer Aggressiveness is Related to Fibronectin Over Expression, Driving the Activation of SDF-1:CXCR4 Axis
Sofia Gouveia-Fernandes1,2, Tânia Carvalho1,2,3, Germana Domingues1,2,3, Renata Bordeira-Carriço1,2, Sérgio Dias1,2,3# and Jacinta Serpa1,2*#
1Centro de Estudos de Doenças Crónicas (CEDOC), Faculdade de Ciências Médicas da Universidade Nova de Lisboa, Portugal
2Unidade de Patobiologia Molecular do Instituto Português de Oncologia de Lisboa Francisco Gentil, EPE (UIPM-IPOLFG), Lisbon, Portugal
3Instituto Gulbenkian de Ciência (IGC), Oeiras, Portugal
#Equal contribution
*Corresponding author:
Jacinta Serpa, Centro de Estudos de Doenças Crónicas (CEDOC), NOVA Medical School/Faculdade de Ciências Médicas, Universidade NOVA de Lisboa Campo Mártires da Pátria 130, 1169-056 Lisbon, Portugal, E-mail: jacinta.serpa@nms.unl.pt
Int J Cancer Clin Res, IJCCR-3-072, (Volume 3, Issue 6), Review Article; ISSN: 2378-3419
Received: August 12, 2016 | Accepted: November 16, 2016 | Published: November 19, 2016
Citation: Gouveia-Fernandes S, Carvalho T, Domingues G, Bordeira-Carrriço R, Sérgio DIAS, et al. (2016) Colorectal Cancer Aggressiveness is Related to Fibronectin Over Expression, Driving the Activation of SDF-1:CXCR4 Axis. Int J Cancer Clin Res 3:072. 10.23937/2378-3419/3/6/1072
Copyright: © 2016 Gouveia-Fernandes S, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Fibronectin (FN), an ECM major component, mediates cells-ECM interactions and has been correlated with cancer progression.
We exploited FN role in colorectal cancer progression. A colon adenocarcinoma cell line (HCT15) over expressing FN (HCT15-FN) was generated and in vitro and in vivo studies were performed.
In vitro, increased proliferation and migration, and decreased apoptosis were observed in HCT15-FN cells, reflecting in vivo bigger tumors, more lung metastases and decreased survival. Disturbing SDF-1:CXCR4 axis by treating mice with AMD3100 prevented disease progression in vivo.
Our findings shed light on the way an ECM component perturbs and favors neoplastic progression, disclosing CXCR4 as a potential target for therapy in colorectal cancer.
Keywords
Fibronectin, Colorectal cancer, SDF-1:CXCR4 axis, Metastasis, Therapeutic target
Introduction
Cancer hallmarks result from dynamical interactions between tumor cells and tumor microenvironment, including normal cells, growth factors and extracellular matrix (ECM) components [1].
Fibronectin (FN) is a high molecular weight, multidomain glycoprotein [2,3], present as a soluble form in body fluids, such as plasma (plasma FN), and as an insoluble form in ECM (cellular FN) [2,4,5]. FN exists, generally, as a dimer, composed of two nearly identical polypeptide chains covalently linked by a pair of C-terminal disulfide bonds. This glycoprotein has been implicated in a wide variety of cell functions, particularly those involving interactions between cells and ECM, through integrin receptors. FN plays important roles in cell adhesion, morphology and spreading, cytoskeletal organization, cell migration, growth and differentiation, phagocytosis, hemostasis and embryonic development [6].
ECM proteolysis is crucial for cancer progression and FN strong susceptibility to proteolytic degradation is well documented [5]. Matrix metalloproteinases (MMPs) represent the most important family of proteases associated to tumorigenesis [7], being MMP-2 and MMP-9 considered the most important in metastasis [5,8,9]. The parallel increase in FN and proteolytic enzymes, together with increased levels in FN fragments is observed in tissues and biological fluids of cancer patients [6,10-12], being related to the stage of disease [10,12]. Given the involvement of FN in cancer cells adhesion to ECM, its over expression influences cancer cells invasion and metastasis [2,3]. In vitro, FN has been shown, in several studies, to induce MMPs expression and to modulate different biological properties of cancer cells [11,13-16].
In contrast, other studies attributed anti-tumor properties, including anti-metastatic activity, to some fragments derived from FN [17,18] .
ECM represents a critical target of oncogenic transformation. Among its most abundant components, FN stands out and recent findings suggest FN may be linked with metastatic capacity of pancreatic cancer [19] and FN controlled synthesis by cancer cells is recognized as a crucial feature of the "pre-metastatic niche" [20], a key event in metastatic cancer spread.
Here, we explored the role of FN in modulating cancer progression using a colorectal in vitro and in vivo model.
Methods
Reverse transcription and polymerase chain reaction (rt-pcr)
RNAs were obtained using RNeasy RNA extraction kit (CatNo-19503-Qiagen) and cDNA was synthesized from 1 μg of RNA, using Superscript II® (CatNo-18064-014Invitrogen), according to manufacturer's protocols.
Construction of pcdna3.1 - full-length fibronectin (pcdna3-flfn)
Full-length FN (embryonic) was amplified from cDNA synthesized from SW480 cells (ATCC, CCL-228™) RNA. cDNA synthesis was performed as described above, but a panel of 23 reverse primers of 15 mer (Supplementary Table 1) was used instead of random primers, to ensure the specificity and enrichment of cDNA from FN; cDNA synthesis occurred for 4 hours. PCR was performed by using an Expand Long Template PCR System (CatNo-11681834001-Roche), according to manufacturer's instructions. Primers were designed as instructed by pcDNA3.1 Directional TOPO Expression kit protocol (CatNo-K4900-40-Invitrogen) (Supplementary Table 1).
The amplified product was visualized in a 1.2% (w/v) agarose gel electrophoresis with 0.005% (v/v) ethidium bromide. DNA fragment with the expected size was extracted using a QIAquick Gel Extraction kit (CatNo-2870-Qiagen), according to manufacturer's protocol.
pcDNA3-flFN expression vector was constructed by using the pcDNA™.1 Directional TOPO Expression kit (CatNo-K4900-40-Invitrogen), and cloned by transforming One-shot TOP 10 Chemically Competent Escherichia coli (Invitrogen), according to the manufacturer's protocol. Plasmids were isolated by using a Plasmid DNA MiniPreps kit (EasySpin), according to manufacturer's instructions.
The plasmid with the expected size was selected by digestion with restriction enzymes (NotI (CatNo-ER0591-Fermentas) and BamHI (CatNo-ER0051-Fermentas)), at 37 °C, and DNA was sequenced (Supplementary Table 1).
Sequencing reactions were performed with BigDye® Terminator v3.1 Cycle Sequencing kit (CatNo-4337456-Applied Biosystems). The automated sequencing was performed in an ABI Prism™ 310 Genetic Analyzer (Applied Biosystems) and analyzed with Sequencing Analysis 3.4.1 software.
Cell culture and stable cells transfection
Cells were maintained at 37 °C in a humidified 5% CO2 environment in MEM Alpha Medium 1X (CatNo-12000-022-Invitrogen), supplemented with 10% Fetal Bovine Serum (FBS) (CatNo-10270-106-Invitrogen) and 1X Antibiotic-Antimycotic (CatNo-15240-062-Invitrogen).
HCT15 cells (ATCC, CCL-225™) were transfected with pCDNA3-flFN, using Lipofectamine™ 2000 (CatNo-11668-019-Invitrogen), according to the manufacturer's instructions. Cells were selected using Geneticin (G418-sulphate) (CatNo-11811-031-Invitrogen). Decreasing concentrations were used each week; the lower concentration was maintained: 1 mg/ml, 500 μg/ml, 100 μg/ml, 50 μg/ml and 10 μg/ml.
Real-time pcr and human metastasis array
Real-time PCR was performed using 1 μl of cDNA, synthesized as described above, and SYBR® Green Master Mix (CatNo-4309155-Applied Biosystems), according to the manufacturer's instructions. RNA 18S was used as housekeeping gene. Samples were analyzed in triplicate. Reactions were developed in ABI Prism® 7900HT (Applied Biosystems).
The Human Tumor Metastasis RT2 Profiler™ PCR Array (CatNo-PAHS-028Z-Qiagen), was performed following the manufacturer's protocol.
Immunofluorescence
Cells were grown on 0.2% gelatin coating and fixed in 2% paraformaldehyde for 15 minutes at 4 °C. Tissue samples were fixed in 10% formalin for 48 hours and embedded in paraffin. Both were blocked with 0.2% (w/v) BSA in PBS 1X for 1 hour at room temperature, and incubated with primary antibody overnight (diluted in 0.1% (w/v) BSA in PBS 1X, 1:100). Antibodies used were: polyclonal anti-FN1−C-Terminal (CatNo-SAB4500974-Sigma-Aldrich); monoclonal anti-α3 integrin (CatNo-MAB1952Z-Millipore); monoclonal anti-β1 integrin (CatNo-EP1041Y-Abcam); polyclonal anti-β3 integrin (CatNo-ab47584-Abcam); anti-human CXCR4 (CatNo-FAB170F-R&D Systems), and anti-CK19 (CatNo-MAB3506-R&DSystems). Samples were incubated with secondary antibody - Alexa Fluor® 488 anti-mouse (CatNo-A-11001-Invitrogen), Alexa Fluor® 488 anti-rabbit (CatNo-A-11008-Invitrogen) and Alexa Fluor® 488 anti-rat (CatNo-A-11006-Invitrogen) - for 2 hours, at room temperature. Slides were mounted in VECTASHIELD media with DAPI (4'-6-diamidino-2-phenylindole) (CatNo-H-1200-Vector Labs) and examined in an Axio Imager.Z1 microscope (Zeiss). Images were acquired with AxioVision software and processed with ImageJ software.
Western blotting
Cell extracts were performed with RIPPA buffer + 1 mM Na2VO3 + 1 × EDTA - free protease inhibitor cocktail (CatNo-S8830-Roche), on ice for 30 min. From each lysate 100 μg of protein were collected and boiled at 95 °C, for 10 minutes, with β-mercaptoethanol:SDS loading buffer 5X (1:10). Proteins were resolved by electrophoresis on 15% SDS–PAGE at 150V, for 1 hour, into TGS buffer 1X (CatNo-161-0732-Bio-Rad), in a Mini-PROTEAN Tetra Electrophoresis System (Bio-Rad), and then transferred to a nitrocellulose membrane (CatNo-162-0115-Bio-Rad), into transfer buffer, at 60V, overnight, at 4 °C. Membrane blocking was performed in a 5% (w/v) solution of non-fat milk in 0.1% (v/v) Tween 20 in PBS 1X.
Primary antibodies used were: monoclonal anti-human Bcl2 (1:1000) (CatNo-SAB4300340-Sigma-Aldrich), polyclonal anti-human Bax (1:100) (CatNo-HPA027878-Sigma-Aldrich) and monoclonal anti-β-actin (1:5000) (CatNo-A5441-Sigma-Aldrich).
The secondary antibodies were anti-rabbit (CatNo-G-21234-Thermo Scientific) and anti-mouse (CatNo-G-21040-Thermo Scientific), conjugated with horseradish peroxidase (HRP). Membrane was developed using SuperSignal® West Pico Chemiluminescent Substrate (CatNo-34077-Thermo Scientific). Bands were quantified using Image J software.
Cell proliferation, cell aggregation, apoptosis and migration
For cell proliferation, 106 cells were plated in T-25 flasks, in triplicate. When adherent, cells were maintained serum free, overnight. Complete growth medium was added and cells were harvested with 0,25%-Trypsin-EDTA (CatNo-25200-056-Invitrogen), for cell number counting and apoptosis evaluation at 0, 3, 6, 24 and 30 hours.
For apoptosis analysis, cells were rinsed with PBS 1X and ressuspended in 0.1% (w/v) BSA in PBS 1X. Cells were incubated with FITC-Annexin V (CatNo-640906-BioLegend) (diluted in annexin binding buffer, 1:40) for 15 minutes, at room temperature. Cells were rinsed with 0.1% (w/v) BSA in PBS 1X and ressuspended in annexin-V binding buffer. Acquisitions were performed using a FACSCalibur (BD Biosciences) and further analyzed with CellQuest Pro software.
Cell aggregation assay was performed in triplicate by coating wells in a 48-well plate with 150 μl of soft-agar solution with subsequent cell seeding (4 × 104 cells per well). Aggregation was evaluated at 24 h under an Olympus CK2 inverted optical microscope (Olympus).
For in vitro wound healing assay, cells were grown in a 3.8 cm2 tissue culture wells to a confluent monolayer. In each well, a scratch was made with a P20 pipette tip to the length of the well. After the scratch, the culture medium was replaced to remove detached cells. A time-lapse experiment was performed and followed under an Olympus CK2 inverted optical microscope (Olympus). Phase images were acquired at the following time points: 0, 8, 24 and 36 hours.
Zymography
Culture supernatants were concentrated by using Amicon® Ultra-4 Centrifugal Filter Units (Millipore). SDS loading buffer 5X was added to each sample and the mixtures were loaded in a 12% polyacrilamide gel with 0.1% (w/v) gelatin. Electrophoresis was performed with a Mini-PROTEAN Tetra Electrophoresis System (Bio-Rad), 150 V, for 90 minutes, into TGS buffer 1X (Bio-Rad). Gel was incubated in renaturating buffer (25% TritonX-100 (v/v)) for 1 hour, with agitation, and then gel was incubated, overnight, 37 °C, in collagenase buffer. Staining was performed with 0.5% (w/v) Coomassie Blue R-250 (CatNo-20278-Thermo Scientific) for 30 minutes and destaining with destilled water, with agitation. The gel was scanned and bands were quantified using Image J software.
in vivo colorectal cancer murine model
in vivo experiments were performed in 6 weeks old BALB/c-SCID male mice.
Cells were rinsed in PBS 1X, harvested by scraping, and ressuspended in PBS 1X. To induce orthotopic xenograft tumors, each mouse was anesthetized by intraperitoneal injection of ketamine/medetomidine (Medetor®-Virbac) and 106 cells were surgically injected in cecum visceral wall. Mice were awakened with atipamezole reversor (Revertor®-Virbac) and infections were prevented with enrofloxacine/meloxicam antibiotic/anti-inflammatory (Baytril®, CatNo08711367-Bayer; Metacam®, CatNo-12301231-Boehringer-Ingelheim) for 3 days. Drug doses were used according to the manufacturers.
Four groups were established: mice injected with control HCT15 with (n = 4) or without CXCR4 antagonist, octahydrochloride hydrate (AMD3100) (CatNo-A5602-Sigma) (n = 4), and HCT15-FN with (n = 4) or without AMD3100 (n = 4). AMD3100 was administered at a dose of 1.25 mg/kg by intraperitoneal injection, each 3 days, starting from day 14 after orthotopic tumor inoculation.
After 2 months, mice were euthanized in a CO2 chamber.
Tumor volume was determined as (minor axis2 × major axis)/2. Tumors and main target organs for metastasis were collected for histological analysis.
Statistics
Data were analyzed using t-tests (two-tailed) with Excel (Microsoft) and GraphPad prism 5. Statistically significant changes were determined at p < 0.05.
Results
Generation of hct15-fn colorectal cancer cell line
To study the role of FN in modulating tumor onset and progression, a colon adenocarcinoma cell line (HCT15) over expressing FN was generated, by stable transfection with human embryonic FN gene (full length FN), pcDNA3-flFN. This cell group was compared to its wild type counterpart (HCT15 control) in in vitro and in vivo assays. As shown in figure 1A, FN-transfected cells over expressed FN at the mRNA level; this was further confirmed at the protein level (Figure 1B). Interestingly, HCT15-FN cells also over expressed integrins alpha3 and beta 3, when compared to HCT15 control cells.
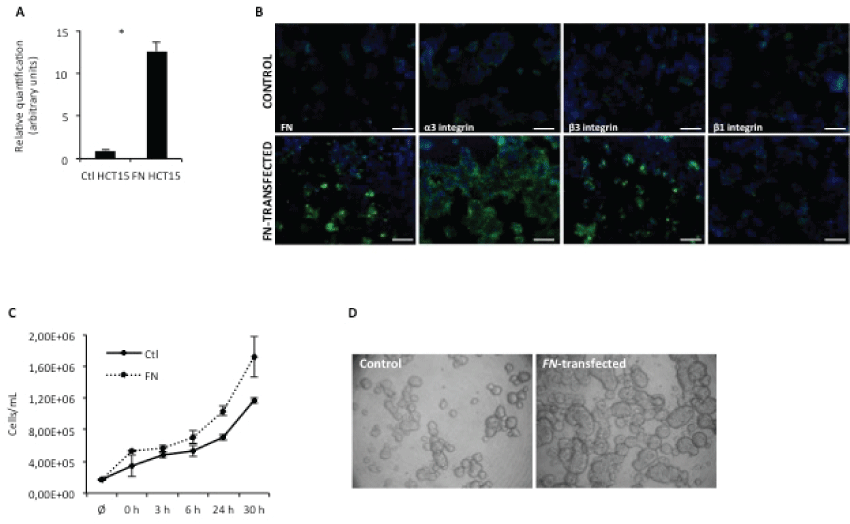
.
Figure 1: FN overexpression increases cell proliferation.
A) Real-time PCR for FN expression. Relative quantification of FN mRNA levels in control and HCT15-FN(* < 0.05); B) Immunoreactivity for FN, α3, β3 and β1 integrins in control and HCT15-FN cells by fluorescence microscopy (original magnification: 200x). Nuclei were stained with DAPI (blue). Scale bar: 100 μm; C) Cell proliferation curve for control and FN-transfected cells at 0, 3, 6, 24 and 30 hours; D) Slow aggregation assay performed in control and FN-over expressing cells at 24 hours. Phase microscopy (original magnification: 100x). Error bars represent standard deviation.
View Figure 1
Fn overexpression results in increased cell proliferation, larger cell aggregates and reduced apoptosis
To evaluate the role of FN over expression in cell proliferation, growth curves were performed, at different time points – 0, 3, 6, 24 and 30 hours (Figure 1C). Data showed HCT15-FN had a higher growth rate than HCT15 control cells.
The effect of FN over expression on cell-cell adhesion was also evaluated, by slow aggregation assay (Figure 1D). An increase in cell-cell adhesion was observed for cells over expressing FN, as well as the formation of larger aggregates than in HCT15 control cells.
Apoptosis was assessed by Bax/Bcl-2 ratios (Figure 2A) and annexin V staining (Figure 2B). Bax (pro-apoptotic) and Bcl-2 (anti-apoptotic) expressions were determined by Western blotting. The results showed a lower Bax/Bcl-2 ratio for HCT15-FN, in comparison to HCT15 control cells, suggesting a lower activation of apoptosis.
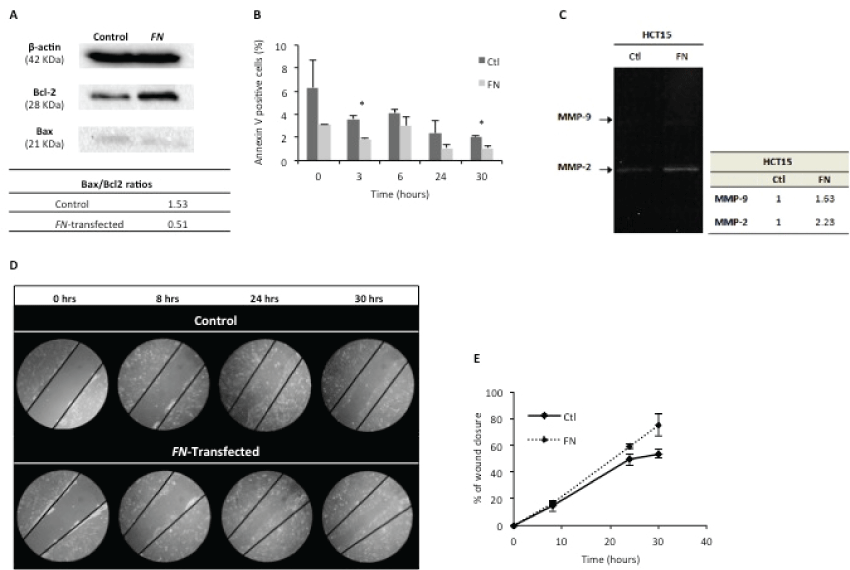
.
Figure 2: FN over expression reduces apoptosis and is associated to a higher cell migration rate.
A) Assignment of Bax/Bcl-2 ratios, by Western blotting, for control and FN-transfected cells. β-actin was used as housekeeping protein. One hundred μg of protein were loaded in each lane. Bands were quantified using ImageJ software; B) Apoptosis analysis by annexin V – 7AAD staining in control and HCT15-FN at 0, 3, 6, 24 and 30 hours (*p < 0.05); C) Zymography of cells media supernatants; D) Wound healing assay for control and transfected cells, at 0, 8, 24 and 30 hours. Phase microscopy (original magnification: 200x); E) Quantification of wound closure, at 0, 8, 24 and 30 hours. Bands were quantified using ImageJ software. Error bars represent standard deviation.
View Figure 2
Annexin V staining was used to quantify the number of cells undergoing apoptosis within each cell group (Figure 2B). Data showed HCT15-FN cells undergo apoptosis at a statistically significant (p < 0.05) lower rate than HCT15 control cells, at 3 and 30 hours time points.
Fn overexpression is associated with higher proteolytic activity and higher migratory capacity of hct15 cells
Another attribute of cancer is the ability of cells to migrate from the original location to invade the surrounding tissues and, ultimately, metastasize at distant organs [21]. During these stages, ECM proteolysis is crucial and MMPs assume an important role in this context [5,7].
FN ability to interfere in MMPs production/secretion and/or activity was assessed by gelatinolytic zymography of cell culture conditioned media (Figure 2C). The results revealed higher enzymatic activity of MMP-9 and MMP-2 in HCT15-FN cells than in HCT15 control cells.
In vitro wound healing assay was performed to assess the role of FN on the migratory rate of cells, by comparing HCT15 control and HCT15-FN (Figure 2D and Figure 2E). HCT15-FN showed a higher migratory capacity than HCT15 control cells.
Fn ovexpression induces cxcr4 and flt-4 expression
We exploited the mechanisms underlying the phenotypes described above, related to FN over expression. For this purpose, an array of genes related to human metastasis was carried out for HCT15 control and HCT15-FN. Data were validated by a second real-time PCR (Figure 3A). Statistically significant (p < 0.05) higher expression of CXCR4, FLT4 and MMP13 and lower expressions of MTA1 and TIMP3 were found for HCT15-FN in comparison to HCT15 control cells.
Given these results, another real-time PCR was carried out targeting CXCR4 and FLT4 ligands – SDF-1 and VEGF C, respectively (Figure 3B). A statistically significant (p < 0.05) increase in SDF-1 and VEGF C expression was also seen in HCT15-FN in comparison to HCT15 control cells.
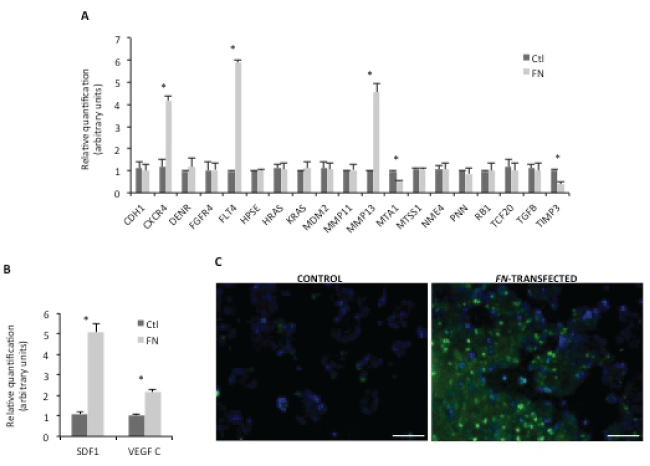
.
Figure 3: Screening for metastasis-related targets.
A) Validation of PCR array for human tumor metastasis by real time PCR. Relative quantification results were compared between control and HCT15-FN (*p < 0.05); B) Real-time PCR for SDF-1 and VEGF C expression. Relative quantification of mRNA levels in control and FN-transfected cells (*p < 0.05); C) Immunoreactivity for CXCR4 in control and FN-transfected cells. Fluorescence microscopy (original magnification: 200x). Nuclei were stained with DAPI (blue). Scale bar: 100 μm. Error bars represent standard deviation.
View Figure 3
Sdf-1:cxcr4 axis blockade reduces tumor progression and metastasis in vivo
Considering the relevance of the SDF-1:CXCR4 axis in cancer progression [22,23], we sought to investigate if this chemokine could be responsible for the overall effects of FN over expression. For this purpose, and after the confirmation of the differential expression of CXCR4, at protein levels (Figure 3C), in vivo experiments were performed to assess the effect of FN over expression on tumor growth and to determine the effect of CXCR4 blockade in the FN-mediated effects. First, the tumorigenic capacity of HCT15 control and HCT15-FN cells was assessed by generating orthotopic xenograft tumors in 6-weeks-old BALB/c-SCID mice. As shown in figure 4A, survival was longer in mice inoculated with HCT15 control, with all animals alive at the end of the experimental period; in contrast, for mice inoculated with HCT15-FN, the overall survival was reduced by 75% at the end of the experiment and mice had to be euthanized. In parallel, we treated mice inoculated with HCT15 control or HCT15-FN cell lines with CXCR4 antagonist-AMD3100. Notably, 100% of mice undergoing AMD3100 treatment survived until the end of the experiment, from HCT15 control and HCT15-FN groups.
Necropsy revealed larger primary tumors in mice inoculated with HCT15 overexpressing FN, when compared to mice inoculated with HCT15 control cells (Figure 4B and Figure 4C). Tumors from mice treated with AMD3100 were however smaller than the respective control group (Figure 4C). Concerning metastasis, histological analysis of liver and lungs revealed the presence of more micro-metastases in mice injected with HCT15-FN cells. In sharp contrast, mice treated with AMD3100, regardless of being inoculated with HCT15 control or HCT15-FN cells, did not develop metastases (Figure 4D).
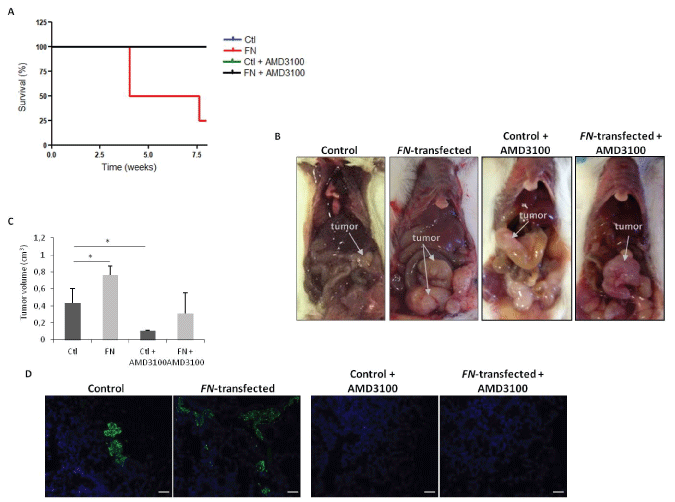
.
Figure 4: Orthotopic xenograft tumors induction of control and HCT15-FN under standard conditions and upon CXCR4 antagonist AMD3100 treatment.
A) Kaplan-Meier survival curves for mice inoculated with control and HCT15-FN, over 8 weeks; B) Macroscopic observation of xenograft orthotopic BALB/c-SCID. Representative images of the animals inoculated with control and FN-transfected cells; C) Primary tumors volumes (*p < 0.05); D) H&E and immunofluorescense (anti-CK19; FITC). Representative images of the lungs of animals inoculated with control and HCT15-FN, not injected and injected with AMD3100, in H&E black arrows indicate metastasis foci. Light and fluorescence microscopy (original magnification: 100x). Scale bar: 50 μm. Error bars represent standard deviation.
View Figure 4
Discussion
ECM represents a critical player in oncogenic transformation. The several steps of cancer are largely dependent on the permissive nature of the microenvironment and ECM proteolysis assumes great importance in this context [24-26]. FN is one of the most abundant components of ECM [2] and several studies related FN levels to tumor progression: in cancer patients, an increase in both FN and FN fragments levels was observed [6,10,11,27] and, in several reports where functional studies on cancer cell lines were performed an equivalent correlation has also been observed [11,13-16] .
In the context of solid tumor growth, colorectal cancer assumes a relevant position as one of the most common and lethal malignancies worldwide [28]. In order to explore the role of FN in tumor development and progression, colon cancer was the chosen model. For this purpose, a stably FN-overexpressing HCT15 cell line was generated, which features were compared to its wild type counterpart (HCT15 control) in in vitro and in vivo assays.
Interestingly, the increase in FN in HCT15-FN cells was accompanied by an increase in α3 and β3 integrins expression, which may suggest a cross-regulatory mechanism, since these integrins are FN receptors [29].
Subsequent detailed characterization of the FN-transfected cell line versus its parental counterpart revealed that the first had a higher proliferative rate, was able to form larger cell aggregates, showed lower levels of apoptosis and increased migratory capacity.
MMP-9 and MMP-2 enzymatic activities were also increased in HCT15-FN, accounting for the increased invasion capacity. Besides the evidence that MMPs activity is strongly associated with tumorigenesis [7], MMP-9 and MMP-2 are thought to be crucial for metastases formation [5,8,9,30]. These increases in MMPs activity might be related to higher amounts of FN in supernatants of HCT15-FN cells, which is cleaved by MMPs [7,11], and/or FN may activate signaling pathways responsible for MMPs regulation, as it has been suggested by some studies using different cancer models [14,15,31-33]. The correlation between FN and MMPs expression was further supported from data obtained by performing a PCR-metastasis array, which showed increased expressions in MMP-13 and tissue inhibitor of metalloproteinases-3 (TIMP-3), which target MMPs including MMPs -2 , -9 and -13 [7].
As mentioned, ECM proteolysis, mainly by MMPs, is one of its most important events accompanying cell migration [34]. Data reported here, showing increased migration and MMP activity by HCT15-FN cells, are therefore in agreement.
Next, we exploited the mechanisms underlying the correlation between FN and further tumor progression, by performing a screening for human metastasis targets. Results revealed an increased expression of CXCR4 and FLT4 and their respective ligands SDF-1 and VEGF C in HCT15-FN in comparison to HCT15 control cells.
Considering the wide spectrum of roles of SDF-1:CXCR4 axis [22,23], we hypothesized this could be an important causal link between increased FN amounts and a more aggressive phenotype in colorectal cancer.
To corroborate the in vitro findings and to validate this assumption, two in vivo experiments were developed in parallel. First, survival of mice inoculated with HCT15-FN was shown to be lower than that of mice injected with parental/control cells. Tumors formed by HCT15-FN cells were also larger than those formed by HCT15 control cells (Figure 4B and Figure 4C); and mice inoculated with HCT15-FN showed more metastases than HCT15 control mice (Figure 4D). These observations are consistent with the in vitro results. Second, CXCR4 blockade was shown to promote survival by reducing tumor growth and metastases formation both in mice inoculated with HCT15 control and, more significantly, in HCT15- FN cells.
Taken together, we assumed that SDF-1:CXCR4 axis emerges as a causal link between increased FN amounts and a more aggressive phenotype in colorectal cancer. A role for SDF-1:CXCR4 has already been suggested in other cancers, including breast [35], lung [36], prostate [37] and pancreatic cancer [38]. CXCR4 expression was also shown to be associated with poor prognosis in colorectal cancer patients [39,40] and with the presence of distant metastases in patients with colorectal cancer, as well as with tumor cells migration in vitro [41,42]. Studies on prostate cancer cell lines also linked CXCR4 to increased cell migration and metastasis, through its regulation of MMPs [43,44] and VEGF [43]. On the other hand, the inhibition of either the expression or function of CXCR4 produced a beneficial (clinical) effect in different neoplasms [35,45-49]. Moreover, the SDF-1:CXCR4 axis has been related to ECM components, namely FN, in malignancy contexts. A study on breast cancer showed the involvement of chemokines in tumor cells migration and motility, attributing an important role of CXCR4 in guiding breast cancer cells to target organs, due to a cross-signaling with integrins in an ECM-dependent way [35]. SDF-1-induced integrin activation, through CXCR4, in small cell lung cancer, also appeared to co-operate in mediating adhesion, namely to FN, and chemoresistance [49]. SDF-1 was also shown to affect the metastasis-related behavior of colorectal cancer cells expressing CXCR4, including an increase in cell proliferation, cell adhesion to FN and cell migration [42].
Conclusions
In conclusion, our findings suggest an active role for FN in promoting colorectal cancer progression and metastases formation, via integrins modulation and by promoting SDF-1:CXCR4 interaction.These studies shed light on the mechanisms whereby an ECM component perturbs and favors neoplastic progression, and reveals novel targets for therapeutic intervention in a setting of invasive colorectal cancer.
Acknowledgments
The authors would like to acknowledge to Instituto Portugues de Oncologia de Lisboa Francisco Gentil, EPE and to Liga Portuguesa Contra o Cancro for supporting the research project. iNOVA4Health - UID/Multi/04462/2013, a program financially supported by Fundacao para a Ciencia e Tecnologia/Ministerio da Educacao e Ciencia, through national funds and co-funded by FEDER under the PT2020 Partnership Agreement is acknowledged.
Conflict of Interest
The authors disclose no conflict of interests.
Author Contributions
Sofia Gouveia-Fernandes: Development of experimental work, analysis and interpretation of data and writing of the manuscript
Tania Carvalho: Development of animal models
Germana Domingues: Development of experimental work
Renata Carrico: Development of experimental work
Sergio Dias: Study supervision and revision of the manuscript
Jacinta Serpa: Conception and design of the project; supervision of experimental work and revision of the manuscript.
References
-
Tlsty TD, Coussens LM (2006) Tumor stroma and regulation of cancer development. Annu Rev Pathol 1: 119-150.
-
Pankov R,Yamada KM (2002) Fibronectin at a glance. J Cell Sci 115: 3861-3863.
-
Kaspar M, Zardi L, Neri D (2006) Fibronectin as target for tumor therap. Int J Cancer 118: 1331-1339.
-
Tamkun JW, Hynes RO (1983) Plasma fibronectin is synthesized and secreted by hepatocytes. J Biol Chem 258: 4641-4647.
-
Klein G, Vellenga E, Fraaije MW, Kamps WA, de Bont ES (2004) The possible role of matrix metalloproteinase (MMP)-2 and MMP-9 in cancer, e.g. acute leukemia. Crit Rev Oncol Hematol 50: 87-100.
-
Labat-Robert J (2002) Fibronectin In Malignancy. Semin Cancer Biol 12: 187-195.
-
Kessenbrock K, Plaks V, Werb Z (2010) Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 141: 52-67.
-
Malemud CJ (2006) Matrix metalloproteinases (MMPs) in health and disease: an overview. Front Biosci 11: 1696-1701.
-
Coussens LM, Fingleton B, Matrisian LM (2002) Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science 295: 2387-2392.
-
Katayama M, Kamihagi K, Nakagawa K, Akiyama T, Sano Y, et al. (1993) Increased fragmentation of urinary fibronectin in cancer patients detected by immunoenzymometric assay using domain-specific monoclonal antibodies. Clin Chim Acta 217: 115-128.
-
Kenny HA, Kaur S, Coussens LM, Lengyel E (2008) The initial steps of ovarian cancer cell metastasis are mediated by MMP-2 cleavage of vitronectin and fibronectin. J Clin Invest 118: 1367-1379.
-
Waalkes S, Atschekzei F, Kramer MW, Hennenlotter J, Vetter G, et al. (2010) Fibronectin 1 mRNA expression correlates with advanced disease in renal cancer. BMC Cancer 10: 503.
-
Ding J, Li D, Wang X, Wang C, Wu T (2008) Fibronectin promotes invasiveness and focal adhesion kinase tyrosine phosphorylation of human colon cancer cell. Hepatogastroenterology 55: 2072-2076.
-
Sen T, Dutta A, Maity G, Chatterjee A (2010) Fibronectin induces matrix metalloproteinase-9 (MMP-9) in human laryngeal carcinoma cells by involving multiple signaling pathways. Biochimie 92: 1422-1434.
-
Moroz A, Delella FK, Lacorte LM, Deffune E, Felisbino SL (2013) Fibronectin induces MMP2 expression in human prostate cancer cells. Biochem Biophys Res Commun 430: 1319-1321.
-
Maity G, Fahreen S, Banerji A, Roy Choudhury P, Sen T, et al. (2010) Fibronectin-integrin mediated signaling in human cervical cancer cells (SiHa). Mol Cell Biochem 336: 65-74.
-
Ming Yi, Erkki Ruoslahti (2001) A fibronectin fragment inhibits tumor growth, angiogenesis, and metastasis. Proc Natl Acad Sci U S A 98: 620-624.
-
Kato R, Ishikawa T, Kamiya S, Oguma F, Ueki M, et al. (2002) A new type of antimetastatic peptide derived from fibronectin. Clin Cancer Res 8: 2455-2462.
-
Yu M, Ting DT, Stott SL, Wittner BS, Ozsolak F (2012) RNA sequencing of pancreatic circulating tumour cells implicates WNT signalling in metastasis. Nature 487: 510-513.
-
Sleeman JP (2012) The metastatic niche and stromal progression. Cancer Metastasis Rev 31: 429-440.
-
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144: 646-674.
-
Burger JA, Kipps TJ (2006) CXCR4: a key receptor in the crosstalk between tumor cells and their microenvironment. Blood 107: 1761-1767.
-
Balkwill F (2004) The significance of cancer cell expression of the chemokine receptor CXCR4. Semin Cancer Biol 14: 171-179.
-
Polette M, Nawrocki-Raby B, Gilles C, Clavel C, Birembaut P (2004) Tumour invasion and matrix metalloproteinases. Crit Rev Oncol Hematol 49: 179-186.
-
Chambers AF, Matrisian LM (1997) Changing views of the role of matrix metalloproteinases in metastasis. J Natl Cancer Inst 89: 1260-1270.
-
Werb Z (1997) ECM and cell surface proteolysis: regulating cellular ecology. Cell 91: 439-442.
-
Warawdekar UM, Zingde SM, Iyer KS, Jagannath P, Mehta AR, et al. ( 2006) Elevated levels and fragmented nature of cellular fibronectin in the plasma of gastrointestinal and head and neck cancer patients. Clin Chim Acta 372: 83-93.
-
Xiang L, Xie G, Ou J, Wei X, Pan F, et al. (2012) The extra domain A of fibronectin increases VEGF-C expression in colorectal carcinoma involving the PI3K/AKT signaling pathway. Plos One 7: E35378.
-
Akiyama SK, Olden K, Yamada KM (1995) Fibronectin and integrins in invasion and metastasis 14: 173-189.
-
Serpa J, Caiado F, Carvalho T, Torre C, Gonçalves LG , et al. (2010) Butyrate-rich colonic microenvironment is a relevant selection factor for metabolically adapted tumor cells. J Biol Chem 285: 39211-39223.
-
Das S, Banerji A, Frei E, Chatterjee A (2008) Rapid expression and activation of MMP-2 and MMP-9 upon exposure of human breast cancer cells (MCF-7) to fibronectin in serum free medium. Life Sci 82: 467-476.
-
Han S, Ritzenthaler JD, Sitaraman SV, Roman J (2006) Fibronectin increases matrix metalloproteinase 9 expression through activation of c-Fos via extracellular-regulated kinase and phosphatidylinositol 3-kinase pathways in human lung carcinoma cells. J Biol Chem 281: 29614-29624.
-
Mitra A, Chakrabarti J, Banerji A, Das S, Chatterjee A (2006) Culture of human cervical cancer cells, SiHa, in the presence of fibronectin activates MMP-2. J Cancer Res Clin Oncol 132: 505-513.
-
Voulgari A, Pintzas A (2009) Epithelial-mesenchymal transition in cancer metastasis: mechanisms, markers and strategies to overcome drug resistance in the clinic. Biochim Biophys Acta 1796: 75-90.
-
Wendel C, Hemping-Bovenkerk A, Krasnyanska J, Mees ST, Kochetkova M (2012) CXCR4/CXCL12 participate in extravasation of metastasizing breast cancer cells within the liver in a rat model. Plos One 7: E30046.
-
Kijima T, Maulik G, Ma PC, Tibaldi EV, Turner RE, et al. (2002) Regulation of cellular proliferation, cytoskeletal function, and signal transduction through CXCR4 and c-Kit in small cell lung cancer cells. Cancer Res 62: 6304-6311.
-
Akashi T, Koizumi K, Tsuneyama K, Saiki I, Takano Y, et al. (2008) Chemokine receptor CXCR4 expression and prognosis in patients with metastatic prostate cancer. Cancer Sci 99: 539-542.
-
Saur D, Seidler B, Schneider G, Algül H, Beck R, et al. (2005) CXCR4 expression increases liver and lung metastasis in a mouse model of pancreatic cancer. Gastroenterology 129: 1237-1250.
-
Saigusa S, Toiyama Y, Tanaka K, Yokoe T, Okugawa Y, et al. (2010) Stromal CXCR4 and CXCL12 expression is associated with distant recurrence and poor prognosis in rectal cancer after chemoradiotherapy. Ann Surg Oncol 17: 2051-2058.
-
Speetjens FM, Liefers GJ, Korbee CJ, Mesker WE, van de Velde CJ, et al. (2009) Nuclear localization of CXCR4 determines prognosis for colorectal cancer patients. Cancer Microenviron 2: 1-7.
-
Schimanski CC, Schwald S, Simiantonaki N, Jayasinghe C, Gönner U,et al. (2005) Effect of chemokine receptors CXCR4 and CCR7 on the metastatic behavior of human colorectal cancer. Clin Cancer Res 11: 1743-1750.
-
Shin HN, Moon HH, Ku JL (2012) Stromal cell-derived factor-1α and macrophage migration-inhibitory factor induce metastatic behavior in CXCR4-expressing colon cancer cells. Int J Mol Med 30: 1537-1543.
-
Wang Q, Diao X, Sun J, Chen Z (2011) Regulation of VEGF, MMP-9 and metastasis by CXCR4 in a prostate cancer cell line . Cell Biol Int 35: 897-904.
-
Singh S, Singh UP, Grizzle WE, Lillard JW Jr (2004) CXCL12-CXCR4 interactions modulate prostate cancer cell migration, metalloproteinase expression and invasion. Lab Invest 84: 1666-1676.
-
Chen Y, Stamatoyannopoulos G, Song CZ (2003) Down-regulation of CXCR4 by inducible small interfering RNA inhibits breast cancer cell invasion in vitro. Cancer Res 63: 4801-4804.
-
Singh B, Cook KR, Martin C, Huang EH, Mosalpuria K, et al. (2010) Evaluation of a CXCR4 antagonist in a xenograft mouse model of inflammatory breast cancer. Clin Exp Metastasis 27: 233-240.
-
Mori T, Doi R, Koizumi M, Toyoda E, Ito D, et al. (2004) CXCR4 antagonist inhibits stromal cell-derived factor 1-induced migration and invasion of human pancreatic cancer. Mol Cancer Ther 3: 29-37.
-
Domanska UM, Timmer-Bosscha H, Nagengast WB, Oude Munnink TH, Kruizinga RC, et al. ( 2012) CXCR4 inhibition with AMD3100 sensitizes prostate cancer to docetaxel chemotherapy 14: 709-718.
-
Hartmann TN, Burger JA, Glodek A, Fujii N, Burger M (2005) CXCR4 chemokine receptor and integrin signaling co-operate in mediating adhesion and chemoresistance in small cell lung cancer (SCLC) cells. Oncogene 24: 4462-4471.
