

Clinical Medical
Reviews and Case Reports
Significance of Antiphospholipid Antibodies in Essential Cryoglobulinemia: A Case Report and Review of Literature
Rama Atluri and Mian Muhammad Rizwan*
Division of Rheumatology, Saint Louis University, St Louis, USA
*Corresponding author: Mian Muhammad Rizwan, Fellow Rheumatology, Division of Rheumatology, 1402 South Grand Boulevard, Doisy Hall, Room 203, St Louis, MO 63104, USA, Tel: 314-977-8832, Fax: 314-977-8818, E-mail: rizwanmm@slu.edu
Clin Med Rev Case Rep, CMRCR-4-151, (Volume 4, Issue 1), Case Report; ISSN: 2378-3656
Received: October 25, 2016 | Accepted: January 11, 2017 | Published: January 13, 2017
Citation: Atluri R, Rizwan MM (2017) Significance of Antiphospholipid Antibodies in Essential Cryoglobulinemia: A Case Report and Review of Literature. Clin Med Rev Case Rep 4:151. 10.23937/2378-3656/1410151
Copyright: © 2017 Atluri R, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Cryoglobulinemia is a rare immune-complex-mediated small vessel vasculitis that has a smoldering clinical course and can potentially involve multiple organ systems. The discovery of its relationship with hepatitis C infection shows the striking association between a viral infection, an autoimmune disease and lymphoproliferative disorders. It is estimated that hepatitis C negative cryoglobulinemia accounts for about 5% to 10% of cases. There have been sporadic reports of association between cryoglobulins and antiphospholipid antibody leading to the suspicion that they participate in the vascular damage at least in some cases but no clear guidelines exist regarding how to best approach such patients if any different at all. We describe a patient with essential cryoglobulinemia, anticardiolipin antibodies hypergammaglobulinemia and take this opportunity to review literature on this topic.
Keywords
Essential mixed cryoglobulinemia, Cryoglobulinemic glomerulonephritis, Antiphospholipid antibodies
Introduction
Mixed cryoglobulinemia (MC) is a complex disorder with a broad spectrum of clinical manifestations. The laboratory hallmark of MC is the presence of cryoglobulins, which are proteins that undergo precipitation upon refrigeration of serum and would dissolve again on warming. To test for them a 20 milliliter blood sample should be stored at 37 °C until completely coagulated, centrifuged at 37 °C at 2500 rpm for 5 minutes, and then stored at 4 °C for 1 week. A white precipitate in the bottom of the tube indicates the presence of cryoglobulins. When the tube is rewarmed to 37 °C, the precipitate will dissolve. They are typically composed of immunoglobulins and complement components. Cryoglobulins are commonly classified into three types as suggested by Brouet, et al. [1]. Type I is composed of isolated monoclonal immunoglobulins (usually of the IgG isotype but less frequently they may be composed of IgM, IgA, or light chains) and is frequently associated with myeloma and Waldenström macroglobulinemia. Type II and type III, designated "mixed cryoglobulinemia", has immuno complexes formed of a monoclonal (type II) or polyclonal (type III) IgM rheumatoid factors (RF) and polyclonal IgG. Type II and type III MC represent up to 80% of all cryoglobulins and are generally present in lower concentrations than monoclonal cryoglobulins. In 1990s it was established that most of these essential MC are associated with chronic hepatitis C virus (HCV) infection [2,3]. We now know that MC is associated with clinical situations that generate large amounts of IgG and immune complexes, including chronic autoimmune diseases such as systemic lupus erythematosus, Sjögren's syndrome [4] or infections such as HCV and HIV infections. MC not associated with those disorders has been defined as essential (primary) MC. The clinical features, etiologies, and treatment of MC not associated with HCV infection have been poorly described.
Cryoglobulins have previously been reported sporadically in association with antiphospholipid antibodies (aPL) antibodies suggesting a possible confounding pathogenic mechanism for vascular injury in such cases [5]. Here we describe a patient who developed cryoglobulinemia and had concurrent elevated levels of autoimmune aPL antibodies.
Case
Our patient was a twenty-eight years old male of Indian inheritance who had no prior medical history. Of note his maternal grandfather and maternal uncle were on hemodialysis for what was presumed to be a complication of hypertension. He went for a routine physical in January 2013. Hyperlipidemia was detected in blood work for which he was started on Atorvastatin. Six months into therapy, he started having muscle pain and cramps that would worsen with intense exercises. He was seen by his primary care provider and was found to have proteinuria and elevated creatine phosphokinase (CPK) on evaluation. A diagnosis of rhabdomyolysis due to statin therapy was made and atorvastatin was discontinued. He continued to have proteinuria on subsequent evaluations and was later referred to nephrologist. Lisinopril was prescribed. He changed his primary doctorin 2014 that again found him to have persistent proteinuria (1.3 gm/day) and now microscopic hematuria on routine screening. The CPK by then had normalized. By then he also started to have sporadic episodes of erythematous, non pruriticpurpura in both legs that would get better in a week or so on its own. He was referred again to nephrologist who did a kidney biopsy and found proliferative glomerulonephritis with membranoproliferative features and large immune complex deposits (Figure 1). Immunofluorescence showed a mixed IgG-IgA cryoglobulins (Figure 2). A strong suspicion of cryoglobulinemic glomerulonephritis was raised on renal biopsy.
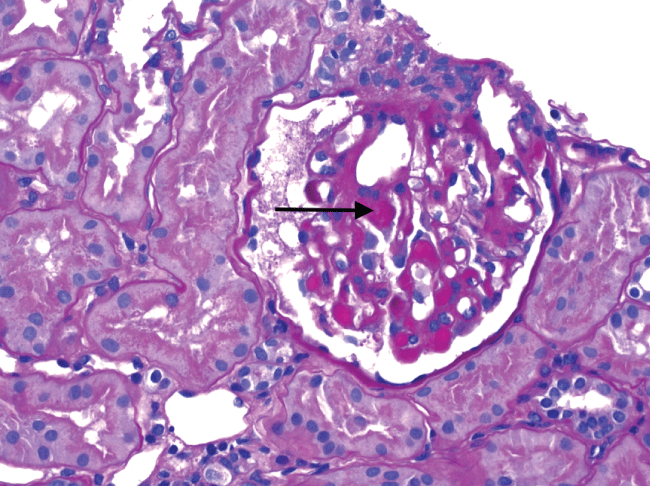
.
Figure 1: Renal biopsy, light microscopy showing PAS positive deposits (arrow) and thrombi.
View Figure 1
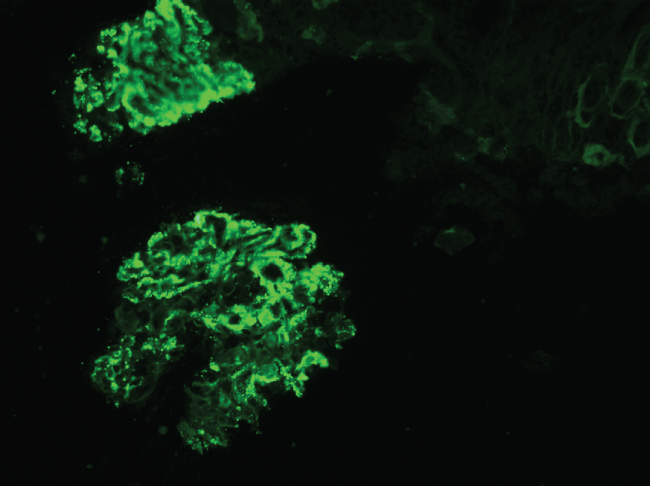
.
Figure 2: Renal biopsy, immunofluorescence images showing IgG granular capillary loop deposits.
View Figure 2
Patient had a positive cryoglobin screen with low cryocet of 1%. His cryoprecipitate contained a monoclonal IgM Kappa and polyclonal IgG globulins the expected composition for MC type II. Patient also had elevated inflammatory markers, positive Rheumatoid factor of 494 IU/ml (Reference < 30 IU/ml), positive lupus anticoagulant screen which was in mildly abnormal range and weakly positive cardiolipin Ab IGMat initially 17 (normal < 12 MPL), 14 and 23.7 upon repeated testing. His IgG anticardiolipin and B2glycoprotien antibodies were negative. Patient also had an incidental finding of Hepatitis A IgG antibody and IgG antibodies against mycoplasma. Immunoglobins analysis showed IGM to be 507 mg/dl (normal 22-292 mg/dl) while IGA and IGM immunoglobins were within normal range. Extensive work up was done including anti chromatin antibody, anti histone antibody, antinuclear antibody, anti Jo antibody, SCL-70 antibody, SSA/SSB antibody, SM/RNP antibody, C3, C4, total complement, ANCA serology with MPO, PR3 and CCP which was all unremarkable. SPEP did not show any evidence of monoclonal protein. Extensive infectious workup including testing for Hepatitis B, C and HIV was unrevealing. His chest x ray did not show any hilar adenopathy. Subsequently he had a flow cytometry, cytogenetics and bone marrow biopsy, which did now show any evidence of malignancy. The presence of high RF and nature of cryocrit supported mixed cryoglobulinemia. Lack of a monoclonal protein ruled out Type 1 cryoglobulinemia. After an extensive infectious and hematology workup we concluded his mixed cryoglobulinemia to be a primary process.
The significance of persistently positive cardiolipin IgM antibody and a weakly positive lupus anticoagulant in absence of any previous clotting events was unclear. He did not meet the laboratory or clinical diagnostic criteria for antiphospholipid antibody syndrome. Our preferred treatment was Rituximab considering his active disease and young age with hopes to preserve his fertility. It was not approved by insurance so he was started on Azathioprine and hydroxychloroquine. While he essentially remains clinically asymptomatic since and his inflammatory markers did improve transiently his cryocet has not changed on therapy. We are in the process of appealing for Rituximab to his insurance. Use of low dose asprin was also suggested to him but he could not tolerate that due to gastrointestinal discomfort.
Discussion
Cryoglobulinemia is an immune-complex-mediated small vessel vasculitis that classically involves the skin, kidneys and peripheral nerves. The cold-induced precipitation of serum proteins was first described in 1933, later Lerner and Watson introduced the term cryoglobulinemia in 1947 [6]. In 1966, Meltzer and Franklin [7] described the typical clinical symptoms associated with cryoglobulinemia, particularly the triad of purpura, arthralgia, and weakness. The frequent finding of liver involvement in such patients which is much less common in other immune complex-mediated diseases, suggested the possibly of a hepatotropic virus playing a part in the disease process. It was not until the 1990s that this was supported by evidence. While the first-generation tests showed antibodies against HCV in 30% to 54% of all patients with MC, with newer tests the prevalence was found to be even higher, ranging from 70% to 100%. It is now estimated that clinical overt cryoglobulinemic syndrome occurs in 15% to 30% of these subjects. The discovery of the relationship between HCV infection and MC shows the striking association between a viral infection, an autoimmune disease and lymphoproliferative disorders. It is estimated that HCV-negative MC accounts for about 5% to 10% of cases [8].
Owing to a smoldering clinical course many features of MC might be overlooked by patients and unwary providers leading to a delay in diagnosis [9]. A nonpruritic, intermittent purpura is often the presenting feature and involves the lower extremities more where venous stasis favors the precipitation of cryoglobulins. Raynaud phenomenon, a monoarticular or oligoarticular intermittent arthritis and peripheral neuropathy have all been reported. Renal involvement is one of the most serious complications of MC and affects approximately one third of patients. Histologically, it almost invariably takes the form of type I membranoproliferative glomerulonephritis with a varying degree of interstitial and vascular damage. If not adequately treated renal failure occurs in 14% of cases. Patients have higher mortality rates than the general population owing to renal involvement and widespread vasculitis (most often involving the gastrointestinal tract). Interestingly higher frequency of RF in cryoglobulinemic patients has also been reported in past which could be related to the RF activity of some cryoglobulinemic component.
One can argue that in some cases perhaps both cryoglobulins and aPL antibodies participate in the vascular damage since there have been sporadic reports of association between cryoglobulins and antiphospholipid antibody. Antiphospholipid antibodies are a heterogeneous group of circulating autoantibodies directed against negatively charged phospholipids and protein cofactors. The presence of aPL antibodies in some but not all individuals confers a risk of a clinical syndrome characterized by recurrent arterial or venous thrombosis, thrombocytopenia, hemolytic anemia, positive Coombs' test, and recurrent idiopathic fetal loss. These aPL, particularly anticardiolipin antibodies (aCL), are found in patients with a wide variety of diseases including autoimmune disturbances, infections, malignancies and in patients receiving drugs known to induce systemic lupus erythematosus. In one study, aCL were present in 35% of patients with primary sjogren's syndrome but not associated with clinical manifestations of antiphospholipid syndrome [4]. These antibodies were usually found with low or moderate titres and are also found to be closely related with hypergammaglobulinemia. The precise pathogenic mechanisms by which this family of autoantibodies might cause vascular injury are still unclear. To understand the impact of prothrombotic conditions on cutaneous vascular pathology it would be worthwhile to review the case of livedoid vasculopathy. Originally described as 'livedoid vasculitis', this disease is characterized by focal purpura progressing to ulceration. Histopathology reveals dermal blood vessel thrombosis and deposition of complement and immunoglobulins, without inflammatory infiltrate or leucocytoclasia [10]. Although literature data are scarce, the histopathologyof ulcerative lesions in cryoglobulinaemia is similar to that of livedoid vasculopathy since thrombosis with minimal inflammation was described at the ulcer edge in a patient with cryoglobulinaemia [11]. This suggests a common pathogenesis for necrotizing cutaneous lesions in mixed cryoglobulinaemia and livedoid vasculopathy. It has also been suggested that while vasculitic damage to the vessel wall initiates the coagulation cascade, it is insufficient to determine tissue necrosis unless a coexisting hypercoagulable state promotes extensive microvascular thrombi formation, eventually leading to necrosis and ulceration. So far few studies have addressed the influence of thrombophilic conditions on the vasculitic manifestations of MC. While an overall 10% prevalence of aCL in patients with HCV-associated cryoglobulinaemia was observed the presence of aCL did not influence the clinical manifestations of cryoglobulinaemia nor was associated with previous episodes of venous or arteriousthromboembolism [12]. It would be interesting to note here that the prevalence of aCL in HCV-infected patients was independent on the presence or absence of cryoglobulinaemia [13] and none of those studies, except one [14] reported vascular complications associated with the presence of aCL. It is also well documented that cryoprecipitation does not interfere in most cases with the measurement of IgM aPL antibody [15]. Lastly, no clear guidelines exist regarding how to best approach such patients if any different at all [16]. It would be interesting to determine the true prevalence of aPL antibodies in patients with cryoglobulinemia, their association with particular clinical manifestations and their role in the pathogenesis of vascular injury in patients with this disease.
Like our patient a relationship between hypergammaglobulinemia and aCL in autoimmune diseases has also been noted in others [4]. The low or moderate titer of aCL seen in most patients, the lack of pathogenicity of these antibodies and the association with hypergammaglobulinemia support a hypothesis that aCL are simply part of the natural repertoire of antibodies in the syndrome characterized with hyper responsive B cells [17].
The treatment of this disorder is still a challenge and ideally should be done by a multidisciplinary team. Efforts should be made to find etiologies other than HCV. The therapeutic management and outcome of infectious forms of cryoglobulinemic vasculitis clearly contrast with that of non-infectious forms [18,19]. The prognosis of non HCV related infectious mixed cryoglobulinemic vasculitis is poor and is mainly related to the causative infectious disease and/or the use of corticosteroids [19]. In HCV-related mixed cryoglobulinemic vasculitis optimal antiviral therapy sometimes associated with corticosteroids is the standard of care for patients with mild to moderate disease activity, whereas rituximab may be used in addition to antiviral agents in patients presenting with severe or refractory disease. The treatment of non-HCV-related MC is usually similar to that of other vasculitides, with steroid as first-line therapy. In non-infectious mixed cryoglobulinemic vasculitis, rituximab plus corticosteroids showed in retrospective studies a greater therapeutic efficacy compared to corticosteroids or and alkylating agents plus corticosteroids to achieve complete clinical, renal and immunological responses [18]. In patients with type I monoclonal cryoglobulinemic vasculitis, the use of alkylating agents, rituximab, thalidomide or lenalinomide, and bortezomib-based regimens represent interesting alternative options [20]. Additional therapies for MC have been proposed, such as imatinib and interleukin-2 inhibitors but future controlled studies are necessary to assess their effectiveness and tolerability in this disease.
Acknowledgment
The authors would like to extend their appreciation for Arkana Laboratories for assistance with histopathology in this case.
References
-
Brouet JC, Clauvel JP, Danon F, Klein M, Seligmann M (1974) Biologic and clinical significance of cryoglobulins. A report of 86 cases. Am J Med 57: 775-788.
-
Dammacco F, Sansonno D, Piccoli C, Tucci FA, Racanelli V (2001) The cryoglobulins: an overview. Eur J Clin Invest 31: 628-638.
-
Shachaf S, Yair M (2014) The correlation between antiphospholipid syndrome and cryoglobulinemia: case series of 4 patients and review of the literature. Revista brasileira de reumatologia.
-
Fauchais AL, Lambert M, Launay D, Michon-Pasturel U, Queyrel V, et al. (2004) Antiphospholipid antibodies in primary Sjögren's syndrome: prevalence and clinical significance in a series of 74 patients. Lupus 13: 245-248.
-
Yancey WB Jr, Edwards NL, Williams RC Jr (1990) Cryoglobulins in a patient with SLE, livedo reticularis, and elevated level of anticardiolipin antibodies. Am J Med 88: 699.
-
Lerner AB, Watson CJ (1947) Studies of cryoglobulins; unusual purpura associated with the presence of a high concentration of cryoglobulin (cold precipitable serum globulin). Am J Med Sci 214: 410-415.
-
Meltzer M, Franklin EC (1966) Cryoglobulinemia--a study of twenty-nine patients. I. IgG and IgM cryoglobulins and factors affecting cryoprecipitability. Am J Med 40: 828-836.
-
Sansonno D, Carbone A, De Re V, Dammacco F (2007) Hepatitis C virus infection, cryoglobulinaemia, and beyond. Rheumatology (Oxford) 46: 572-578.
-
Gheita TA, Khairy NA, Nasrallah MM, Hussein H (2012) Subclinical renal involvement in essential cryoglobulinemic vasculitis and classic polyarteritis nodosa. Joint bone spine 279: 274-280.
-
Hairston BR, Davis MD, Pittelkow MR, Ahmed I (2006) Livedoid vasculopathy: further evidence for procoagulant pathogenesis. Arch Dermatol 142: 1413-1418.
-
Krunic AL, Medenica MM, Laumann AE, Shaw JC (2003) Cryoglobulinaemic vasculitis, cryofibrinogenaemia and low-grade B-cell lymphoma. Br J Dermatol 148: 1079-1081.
-
Casato M, Carlesimo M, Francia A, Timarco C, Antenucci A, et al. (2008) Influence of inherited and acquired thrombophilic defects on the clinical manifestations of mixed cryoglobulinaemia. Rheumatology (Oxford) 47: 1659-1663.
-
Sthoeger ZM, Fogel M, Smirov A, Ergas D, Lurie Y, et al. (2000) Anticardiolipin autoantibodies in serum samples and cryoglobulins of patients with chronic hepatitis C infection. Ann Rheum Dis 59: 483-486.
-
Prieto J, Yuste JR, Beloqui O, Civeira MP, Riezu JI, et al. (1996) Anticardiolipin antibodies in chronic hepatitis C: implication of hepatitis C virus as the cause of the antiphospholipid syndrome. Hepatology 23: 199-204.
-
Bardin N, Pommier G, Sanmarco M (2007) Can cryoglobulins interfere with the measurement of IgM antiphosphatidylethanolamine antibodies by ELISA? Thromb Res 119: 441-446.
-
Hanly JG, Smith SA (2000) Autoimmune antiphospholipid antibodies and cryoglobulinemia. Lupus 9: 264-270.
-
Halse A, Tengnér P, Wahren-Herlenius M, Haga H, Jonsson R (1999) Increased frequency of cells secreting interleukin-6 and interleukin-10 in peripheral blood of patients with primary Sjögren's syndrome. Scand J Immunol 49: 533-538.
-
Terrier B, Krastinova E, Marie I, Launay D, Lacraz A, et al. (2012) Management of noninfectious mixed cryoglobulinemia vasculitis: data from 242 cases included in the CryoVas survey. Blood 119: 5996-6004.
-
Terrier B, Marie I, Lacraz A, Belenotti P, Bonnet F, et al. (2015) Non HCV-related infectious cryoglobulinemia vasculitis: Results from the French nationwide CryoVas survey and systematic review of the literature. J Autoimmun 65: 74-81.
-
Terrier B, Karras A, Kahn JE, Le Guenno G, Marie I, et al. (2013) The spectrum of type I cryoglobulinemia vasculitis: new insights based on 64 cases. Medicine (Baltimore) 92: 61-68.
