

Clinical Medical
Reviews and Case Reports
Adult Rasmussen's Encephalitis
Campanille Veronica1*, De Aguirre Ines2, Crespo Jose1, Gonzalez Macarena1, Lopez Juan Ignacio1, Lucero Carolina1, Perez Garcia Jorgelina1 and Knorre Eduardo1
1Neurology Department, Dr. Teodoro Álvarez Hospital, Argentina
2Instituto de Investigaciones Cardiologicas Dr. Alberto Taquini, Argentina
*Corresponding author: Dr. Veronica Campanille, Servicio de Neurologia, Hospital Teodoro Alvarez Aranguren 2701, Ciudad Autonoma de Buenos Aires, Argentina, E-mail: vcamp@fibertel.com.ar
Clin Med Rev Case Rep, CMRCR-3-144, (Volume 3, Issue 12), Case Report; ISSN: 2378-3656
Received: October 07, 2016 | Accepted: November 28, 2016 | Published: December 01, 2016
Citation: Veronica C, Ines DA, Jose C, Macarena G, Ignacio LJ, et al. (2016) Adult Rasmussen's Encephalitis. Clin Med Rev Case Rep 3:144. 10.23937/2378-3656/1410144
Copyright: © 2016 Veronica C, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Introduction: Rasmussen's Encephalitis (RE) is defined by the presence of partial seizures associated to focal progressive cortical atrophy and in advanced stages hemiparesis, hemianopia and cognitive impairment, though it may appear focal neurological deficits at onset; and it's more prevalent in childhood. Approximately, 10% presents at adulthood. Because of its immunologic physiopathology, RE is considered autoimmune epilepsy. Treatment is focused on antiepileptic drugs (AEDs), steroids, intravenous immunoglobulins (IVIg) and immunosuppressant drugs.
A 37-year-old man presented to the Neurology department in 2015 with a history of epilepsy. Epilepsy began at age 13 with partial motor seizures. In 2011, he had a partial status epilepticus lasting 24 hours and then continued with multiple daily seizures. He had multiple AEDs schemes without good therapeutic response. At admission he had a frequency of 8 seizures in 2 hours. Electroencephalography (EEG), Video electroencephalography (VEEG) and brain magnetic resonance images (MRI) were performed. EEG and VEEG showed left and right partial motor seizures beginning at right frontal lobe. MRI showed mostly right frontal-temporal atrophy and mild involvement of left hemisphere, marking a progression in comparison with previous MRI studies. Cerebrospinal fluid (CSF) analysis indicated hyper proteinorachie, cell count and glucose level were normal. Oligoclonal bands, antineuronal antibodies, rheumatologic and infectious screening were normal. A neuropsychological evaluation was performed, showing mild cognitive impairment, with subcortical dysfunction. The patient received IVIg and there was a transient decrease of seizure frequency of 80%.
Conclusion: This case shows that RE is a progressive and chronic disease, with poor therapeutic response and drug-resistant seizures.
Keywords
Rasmussen's encephalitis, Partial status epilepticus, Drug-resistant autoimmune epilepsy
Introduction
Rasmussen's Encephalitis (RE) is a sporadic, severe cortical and subcortical disease -probably acquired- which is characterized by intractable motor seizures, mainly focal seizures, epilepsia partial continua (EPC) and progressive cognitive impairment with hemiparesis and language and cognitive disorders [1]. In 1958, Rasmussen et al described 3 patients suffering from focal seizures associated with localized chronic encephalitis [2]. It was first described in childhood and associated with viral encephalitis. Later, it was also described in adulthood and other possible etiologies were proposed. RE is a rare disease, and there are only 200 case reports analyzed in current literature. The median age of onset is 1-10 years and it is very infrequent in adolescence and adulthood [3]. There is no sex preference. A viral etiology was already suggested by Rasmussen based on the constituents of the immune reaction in the brains such as lymphocyte infiltration and microglial nodules [4]. However, so far all attempts to identify a pathogenic viral agent have been contradictory and inconclusive. Available data continue to suggest an immune basis to the pathogenesis of RE. Experiments on rabbits demonstrated that antibodies against glutamate/AMPA subunit 3 receptor (GluR3) provoked a clinical response similar to RE [5]. However, these antibodies were not found in all patients with RE; furthermore, anti-GluR3 was also found in other epileptic syndromes [6,7], though some of these reports are not well characterized [8]. Future antibody research in RE will probably concentrate on detecting possibly pathogenic antibodies other than anti-GluR3 [9,10]. A genetic origin has also been suggested, encephalopathy due to focal epilepsy or focal epilepsy due to focal immune response [11]. RE etiology remains unclear, though an immune basis with a role of humoral and cytotoxic response is the most accepted hypothesis.
Disease sets in otherwise healthy children with different types of seizures, especially partial motor seizures including EPC; usually starting in distal limbs. Later, seizures worsen, becoming more irregular and frequent, usually with postictal hemiplegia. In the next stage of the disease, well-stablished deficits appear, with progression over the course of RE. Finally, RE stabilizes with lower seizures but deficits remain [1,12,13].
A special mention is made about the setting of the disease in teenagers and young adults [14], though it has been also described in patients up to age 54 [15]. In this group of patients -which is supposed to represent 10% in some case series [3,16], the course of the disease is usually slower, with less damage and occipital lobe seizures [3]. Furthermore, very few cases of bilateral RE have been described [17,18], as well as others cases starting with hemiparesis and later epilepsy [19], or even cases with unilateral movement disorders, including hemiathetosis and hemidystonia [20].
According to brain biopsies and necropsies there are 4 different pathogenic patterns: group 1, earlier cases show an inflammatory pattern with numerous microglial nodules with or without neuronophagia, perivascular balloon cells and gliosis; group 2 is characterized by microglial nodules, perivascular balloon cells, and some portions of gyral cortex with full necrosis; group 3, neuronal loss and gliosis with extensive perivascular balloon cells and some microglial nodules; and group 4, in late stages, shows few or none microglial nodules, neuronal loss and moderate perivascular inflammation combined with different degrees of gliosis [21]. It was demonstrated that perivascular balloon cells infiltrates were mostly T lymphocytes [22].
According to the 2005 European Consensus, a RE diagnosis can be made by fulfilling three criteria of part A, or two out of three criteria of part B. First, it must be evaluated part A, otherwise part B must be evaluated. Part A: 1. Focal seizures (with or without EPC) and unilateral cortical deficit, 2. EEG showing unihemispheric slowing with or without epileptiform activity and unilateral seizure onset, 3. unihemispheric focal cortical atrophy and at least one of the following: grey or white matter T2/FLAIR hyperintense signal, and hyperintense signal or atrophy of the ipsilateral caudate head. Part B: 1. Epilepsia partial continua or progressive unilateral cortical deficits, 2. progressive unihemispheric focal cortical atrophy, 3. T cell dominated encephalitis with activated microglial cells (typically, but not necessarily forming nodules) and reactive astrogliosis [14].
Evaluation through functional neuroimaging such as positron emission tomography (PET), single photon emission tomography (SPECT) or spectroscopy magnetic resonance imaging (sMRI) has been performed in diagnosis and follows up. Findings described are interictal hypoperfusion and hypometabolism, especially in rolandic cortex and numerous ictal hypometabolic foci. These findings correlate to worst outcome if they appear before structural abnormalities are seen in conventional MRI [23].
Up to date, there haven't been found disease markers to confirm diagnosis. It has been proposed the detection of GlutR3- antibodies in serum but results are controversial [6]. CSF studies show in early stages an unspecific mild increase in proteins and cell count [24]; oligoclonal bands have been found in about 0-67% of patients [25-27].
Numerous treatments with AEDs have been unsuccessful in RE; only limited control on focal seizures and epilepsia partial continua in particular tends to be refractory to antiepileptic drugs.No AEDs alone or in combination show effectiveness. Of note, no data indicating higher efficacy of new AEDs in RE compared to the older ones. As a general rule, number and dose of AEDs should be kept as low as possible, one should try to abolish secondarily generalized tonic-clonic and, possibly, complex partial seizures; EPC, however, is almost never suppressed by AEDs and it provides little benefit to the patients if one tries to suppress this focal motor status epilepticus [28].
Since the first descriptions of the disease, surgery still remains as the most effective treatment for the seizures caused by RE. There have been many surgical approaches but the ones which have been more successful are those related to hemispherectomy -either anatomical or functional. In this respect, hemispherectomy and its modern variants have been found to be the so far and highly effective therapy to achieve seizure freedom. They have fewer complications than anatomical hemispherectomy and produce similar outcomes if they are well performed [29]. Most surgeries have stopped seizures in more than 70 per cent of patients [30,31].
Recently, there have been tried long-term treatments with corticosteroids [17,27,32], treatments with Intravenous Immunoglobulin (IVIG) [32], treatments with plasma-exchange or A protein immune adsorption [27] and Tacrolimus [33]. Only few patients have been treated with Rituximab as an alternative therapy in RE [34].
Case Report
A 37-year-old male patient who is derived from another Institution in 2015. No personal history is presented. His perinatal period as well as his neurologic development has been normal. He works as a private security agent.
History of the actual disease: His epilepsy began at the age of 13. The patient was initially treated with carbamazepine. He remained free of seizure activity for two years. Carbamazepine was prescribed until he was 17-year-old. At this age (1997) he was hospitalized for the increase in seizure frequency and a loading dose of phenytoin was administered. From age 17 to 33 he was treated with valproate, clonazepam, clobazam, topiramate, phenobarbital, phenytoin. As seizure frequency worsen medication dose needed to be adjusted with laboratory tests. He had left sided hemiparesis affecting his face (Todd´s phenomenon) and dysarthria for fifteen minutes. His medical history records (at the age of 16): sharp waves over the right frontal region. Continued left simple partial hemifacial seizures accompanied occasionally by unilateral weakness affecting arms and legs. He couldn't recall having been free of seizure activity during this period. In 2008 at the age of 30 he underwent a MRI. In 2011 (aged 33) a new MRI was performed because of partial continua status epilepticus during approximately 24 hours. From 2011 to 2015: as an outpatient he was treated with multiple drugs: oxcarbazepine: 3600 mg/day, lorazepam: 5 mg/day and levetiracetam: 2000 mg/day. He continued to develop partial seizures. From 2011 onwards the seizure frequency increased to several times a day. In June 2015, on examination at the time of presentation, those seizures persisted without any clinical stability, 20 seizures/day so he was hospitalized. General physical examination was unremarkable except for a moderate dysarthria. VEGG showed: 8 seizures in 2 hours. Laboratory tests were completed (blood and CSF; table 1), VEEG and a new MRI. Routine lab parameters were normal. Antinuclear bodies were negative as well as ADNA, ANA, oligoclonal bands, HIV, PCR of HSV, CMV and EBV. Neurocognitive evaluation revealed a moderate subcortical dysfunction.
![]()
Table 1: Lumbar puncion results. Blood glucose level 108 mg/dl.
View Table 1
Video EEG
Background rhythm during awake stage shows a theta rhythm, 5-6 Hz. Average amplitude, symmetric and synchronic in the posterior regions, it blocks with eye opening. Slow waves in the frontal regions during the entire study. Seizures last 60 seconds. They begin with rhythmic activity in left frontal region at 4 Hz (Figure 1A and Figure 1B), then 3 Hz and later 2 Hz (Figure 1A, Figure 1B and Figure 1C). The patient shows jerks first in the right cheek at a regular interval in 2-4 per second, later right eyelid jerks are observed.
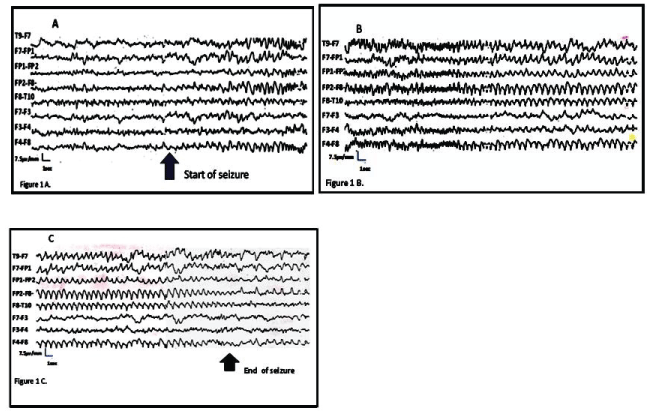
.
Figure 1: Seizures last 60 seconds. 1A,1B) They begin with rhythmic activity in left frontal region at 4 Hz; 1A,1B,1C) then 3 Hz and later 2 Hz; (The patient shows jerks first in the right cheek at a regular interval in 2-4 per second, later right eyelid jerks are observed.
View Figure 1
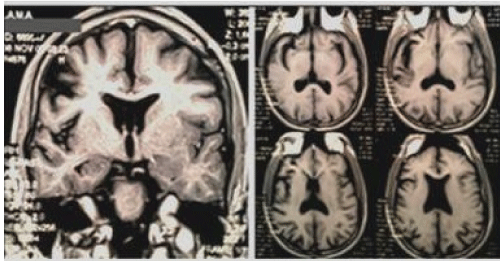
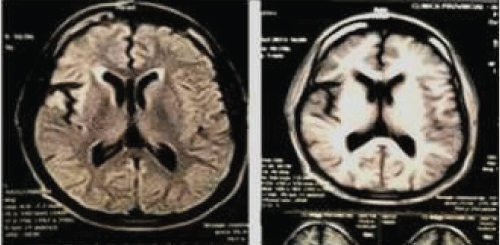
Neuroimaging
Figure 2, figure 3 and figure 4.
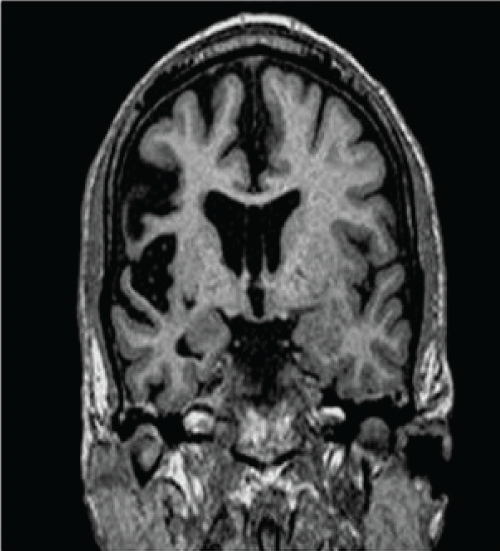
.
Figure 4: Brain MRI 2015. Focal atrophy, unilateral enlargement of the ventricular cisterns. Hyperintensity in FLAIR/T2, especially in the right perisylvian area. Lpsilateral atrophy of caudatus (It might be an early sign). Some compromise of contralateral hemisphere can be seen, probably due to axonal degeneration.
View Figure 4
Treatment
Intravenous Immunoglobulin: 2 g/kg/day fractionated in 5 days, once a month during 5 months. VEEG control: 10 seizures in 4 hours: estimated 62.5% reduction within the ensuing three months after the cycles application; then seizures continued with the same frequency.
Discussion
Rasmussen's encephalitis is produced by chronic progressive inflammation of the brain, with infiltration of T lymphocytes into the brain parenchyma. This inflammation usually affects only one hemisphere of the brain, right or left. It also produces a permanent damage of the nerve cells, leading to atrophy of the affected cerebral hemisphere. The epilepsy caused by the inflammation can also contribute to brain damage. The disease occurs mainly in children and adolescents and rarely in adults.
This case describes an adult patient affected by drug-resistant or refractory epilepsy to different treatment schemes with AEDs, as previous reported in the literature [35]. The type of epilepsy falls into the category of probably symptomatic. The patient developed epilepsia partial continua with focal atrophy, unilateral enlargement of the ventricular system, hyperintensity on FLAIR and T2W, mainly in right perisylvian zone. Ipsilateral atrophy of the head of the caudate nucleus (it can be an early sign). It can be observed compromise of the contralateral hemisphere, probably as a result of degeneration of commissural fibers. Serial MRIs from 2008 to 2015 show progression of atrophy and signal change.
According to Video-EEG this patient has epilepsia partial continua and experiences myoclonus in left sided face, sometimes left leg and right eyelid. He doesn´t have a history of any disease related to CNS. The only explanation for the progressive is the persistence of partial motor seizures which would probably originate an autoimmune response that would in turn play a role in the perpetuation of seizures and therefore disease progress.
The findings of progressive brain atrophy and/or alterations of brain structure based on MRIs, particularly a MRI with gadolinium in a patient with EPC fulfilled the 2005 European consensus diagnosis criteria for the diagnosis of RE [14,36-39].
The patient has refractory epilepsy to medication and progressive focal seizures which have evolved into epilepsia partial continua and significant focal motor-cognitive decline. According to serial neuroimaging he fulfills part A criteria.
The increased protein found in CSF has low specific value because it may be seen in numerous inflammatory, infectious and neoplastic diseases.
The first line treatment proposed is immunotherapy: IV corticosteroids vs IVIG and tacrolimus. This treatment delays surgery, as it has been stablished the only curative treatment of RE. Other options are plasma-exchange, natalizumab, azathioprine and AEDs. Long-term treatment with IVIG may be useful, probably to modify the course of the disease and must be considered prior to surgery [40]. Though surgery is curative, its adverse effects are significant, such as functional deficits. In patients that reached the final stage of RE with minimal functional capacity of the affected hemisphere, surgery is a valid option, especially when seizures became intractable. Corticosteroid therapy results are variable. In a study, the type of epilepsy and motor and cognitive outcome were evaluated in 49 patients of age 8.7 ± 10.5 with RE, who were treated with immunotherapy: IVIG at 100 mg/kg/day, methylprednisolone at 30 mg/kg/day for three days and tacrolimus at initial dose of 0.1 mg/kg/day; compared to functional hemispherectomy. The results were: seizure freedom in 23% with IVIG therapy, 81% with methylprednisolone, 42% with tacrolimus and 71% with surgery. The > 80 intelligence quotients (IQs) were: 43% with IVIG, 50% with corticosteroids, 29% tacrolimus and 0 % with surgery. Only patients treated with immunotherapy showed motor improvement. Hemispherectomy worsened the motor functions in all patients; compared with worsened motor function of 62% of IVIG and 10% of steroids [41,42]. The study made by Bien et al about tacrolimus vs. IVIG showed that both treatments may delay tissue loss and functional deterioration in patients with RE [42].
Plasma-exchange is a useful adjuvant therapy in patients with RE, specifically in those with status epilepticus before surgical removal [11].
Our patient received 5 monthly cycles of IVIG, and there were no modifications in his antiepileptic treatment. The was an estimated seizure reduction of 62.5% during the three months after IVIG therapy. However, after this time, seizures continued with the initial frequency. This patient was not a good candidate for surgery because he had both hemispheres compromised, especially the right lobe and he was right-hand, and it would worsen his quality of life and outcome.
Conclusion
This case of adult RE shows a chronic, progressive disease with drug-resistant seizures. The disease went on a prolonged course compared with children onset, and it had fewer structural lesions. Though RE outcome depends on the underlying etiology, in this case the seizure frequency worsened the course of the disease.
Special thanks to Ms. Elma Cabrera, Video-EEG technician on the making of the registry.
References
-
Panayiotopoulos CP (2005) The Epilepsies: Seizures, Syndromes and Management. Bladon Medical Publishing, Oxfordshire, 207-215.
-
Rasmussen T, Olszewski J, Lloyd-Smith D (1958) Focal seizures due to chronic localized encephalitis. Neurology 8: 435-445.
-
Hart YM, Andermann F, Fish DR, Dubeau F, Robitaille Y, et al. (1997) Chronic encephalitis epilepsy in adults and adolescents: a variant of Rasmussen´s syndrome? Neurology 48: 418-424.
-
Atkins MR, Terrell W, Hulette CM (1995) Rasmussen's syndrome: a study of potential viral etiology. Clin Neuropathol 14: 7-12.
-
Rogers SW, Andrews PI, Gahring LC, Whisenand T, Cauley K, et al. (1994) Autoantibodies to glutamate receptor GluR3 in Rasmussen's encephalitis. Science 265: 648-651.
-
Wiendl H, Bien CG, Bernasconi P, Fleckenstein B, Elger CE, et al. (2001) GluR3 antibodies: Prevalence in focal epilepsy but no specificity for Rasmussen's encephalitis. Neurology 57: 1511-1514.
-
Mantegazza R, Bernasconi P, Baggi F, Spreafico R, Ragona F, et al. (2002) Antibodies against GluR3 peptides are not specific for Rasmussen's encephalitis but are also present in epilepsy patients with severe, early onset disease and intractable seizures. J Neuroimmunol 131: 179-185.
-
Watson R, Jiang Y, Bermudez I, Houlihan L, Clover L, et al. (2004) Absence of antibodies to glutamate receptor type 3 (GluR3) in Rasmussen encephalitis. Neurology 63: 43-50.
-
Lang B, Watson R, Bermudez I, Sattelle D, Jepson J, et al. (2004) Antibodies to neuronal alpha7 acetylcholine receptor in patients with Rasmussen's encephalitis. J Neuroimmunol 154: 192.
-
Whitney KD, Andrews JM, McNamara JO (1999) Immunoglobulin G and complement immunoreactivity in the cerebral cortex of patients with Rasmussen's encephalitis. Neurology 53: 699-708.
-
Andrews PI, Dichter MA, Berkovic SF, Newton MR, McNamara JO (1996) Plasmapheresis in Rasmussen's encephalitis. Neurology 46: 242-246.
-
Oguni H, Andermann F, Rasmussen TB (1992) The syndrome of chronic encephalitis and epilepsy. A estudy based on the MNI series of 48 cases. Adv Neurol 57: 419-433.
-
Andermann F Ed (1991) Chronic encephalitis and epilepsy: Rasmussen´s syndrome. Butterworth- Heinemann, Boston.
-
Bien CG, Granata T, Antozzi C, Cross JH, Dulac O, et al. (2005) Pathogenesis, diagnosis and treatment of Rasmussen encephalitis. A European consensus statement. Brain 128: 454-471
-
Vadlamudi L, Galton CJ, Jeavons SJ, Tannenberg AE, Boyle RS (2000) Rasmussen's syndrome in a 54 year old female: more support for an adult variant. J ClinNeurosci 7: 154-156.
-
Oguni H, Andermann F, Rasmussen TB (1991) The natural history of the syndrome of chronic encephalitis and epilepsy: a study of the MNI series of fortyeight cases. In: Andermann F, Chronic encephalitis and epilepsy. Rasmussen's syndrome. Butterworth-Heinemann, Boston, 7-35.
-
Chinchilla D, Dulac O, Robain O, Plouin P, Ponsot G, et al. (1994) Reappraisal of Rasmussen's syndrome with special emphasis on treatment with high doses of steroids. J NeurolNeurosurg Psychiatry 57: 1325-1333.
-
Tobias SM, Robitaille Y, Hickey WF, Rhodes CH, Nordgren R, et al. (2003) Bilateral Rasmussen encephalitis: post-mortem documentation in a five- year-old. Epilepsia 44: 127-130.
-
Korn-Lubetzki I, Bien CG, Bauer J, Gomori M, Wiendl H, et al. (2004) Rasmussen encephalitis with active inflammation and delayed seizures onset. Neurology 62: 984-986.
-
Bhatjiwale MG, Polkey C, Cox TC, Dean A, Deasy N (1998) Rasmussen's encephalitis: neuroimaging findings in 21 patients with a closer look at the basal ganglia. Pediatr Neurosurg 29: 142-148.
-
Robitaille Y (1991) Neuropathologic aspects of chronic encephalitis. In: Andermann F, Chronic encephalitis and epilepsy. Rasmussen's syndrome. Butterworth-Heinemann, Boston, 79-110.
-
Farrell MA, Droogan O, Secor DL, Poukens V, Quinn B, et al. (1995) Chronic encephalitis associated with epilepsy: immunohistochemical and ultrastructural studies. Acta Neuropathol Berl 89: 313-321.
-
Lee JS, Juhasz C, Kaddurah AK, Chugani HT (2001) Patterns of cerebral glucose metabolism in early and late stages of Rasmussen's syndrome. J Child Neurol 16: 798-805.
-
Rasmussen T, Andermann F (1989) Update on the syndrome of 'chronic encephalitis' and epilepsy. Cleve Clin J Med 56: S181-184.
-
Dulac O, Robain O, Chiron C, Plouin P, Pinel JF, et al. (1991) High-dose steroid treatment of epilepsiapartialis continua due to chronic focal encephalitis. In: Andermann F, Chronic encephalitis and epilepsy. Rasmussen's syndrome. Butterworth-Heinemann, Boston, 193-199.
-
Grenier Y, Antel JP, Osterland CK (1991) Immunologic studies in chronic encephalitis of Rasmussen. In: Andermann F, Chronic encephalitis and epilepsy. Rasmussen's syndrome. Butterworth-Heinemann, Boston, 125-134.
-
Granata T, Gobbi G, Spreafico R, Vigevano F, Capovilla G, et al. (2003) Rasmussen's encephalitis: early characteristics allow diagnosis. Neurology 60: 422-425.
-
Bien CG, Schramm J (2009) Treatment of Rasmussen encephalitis half a century after its initial description:Promising prospects and a dilemma. Epilepsy Research 86: 101-112.
-
Cook SW, Nguyen ST, Hu B, Yudovin S, Shields WD, et al. (2004) Cerebral hemispherectomy in pediatric patients with epilepsy: comparison of three techniques by pathological substrate in 115 patients. J Neurosurg Spine 100: 125-141.
-
Honavar M, Janota I, Polkey CE (1992) Rasmussen's encephalitis in surgery for epilepsy. Dev Med Child Neurol 34: 3-14.
-
Delalande O, Bulteau C, Dellatolas G, Fohlen M, Jalin C, et al. (2007) Vertical parasagittal hemispherotomy: surgical procedures and clinical long-term outcomes in a population of 83 children. Neurosurgery 60: ONS19-32.
-
Hart YM, Cortez M, Andermann F, Hwang P, Fish DR, et al. (1994) Medical treatment of Rasmussen's syndrome (chronic encephalitis and epilepsy): effect of high-dose steroids or immunoglobulins in 19 patients. Neurology 44: 1030-1036.
-
Bien CG, Gleissner U, Sassen R, Widman G, Urbach H, et al. (2004) An open study of tacrolimus therapy in Rasmussen encephalitis. Neurology 62: 2106-2109.
-
Thilo B, Stingele R, Knudsen K, Boor R, Bien CG, et al. (2009) A case of Rasmussen encephalitis treated with rituximab. Nat Rev Neurol 5: 458-462.
-
Carmona-Vazquez Carlos Raúl, Peña-Landín Dora Maricela, Venzor-Castellanos Juan Pablo, Pasquel García Velarde Pedro Mario, Dávila-Gutiérrez Guillermo (2014) Encefalitis de Rasmussen: Complejidad del manejo de una epilepsia potencialmente farmacorresistente ilustrada por dos casos clínicos. Rev Mex Neuroci Marzo 15: 119-124.
-
Bauer G, Gotwald T, Dobesberger J, EmbacherN, Felber S, et al. (2006) Transient and permanent magnetic resonance imaging abnormalities after complex partial status epilepticus. Epilepsy Behav 8: 666-671.
-
Villalobod-Chávez JJ, Rodriguez-Uranga G, Sanz-Fernandez (2005) Cambios Secuenciales en la resonancia magnética en estado epiléptico límbico. Rev Neurol 40: 354-357.
-
Amato C, Elia M, Musumeci SA, Bisceglie P, Moschini M (2001) Transient MRI abnormalities associated with partial status epilepticus: a case report. Eur J Radiol 38: 50-54.
-
Sirven JI, Zimmerman RS, Carter JL, Drazkowski JF, Larson JS (2003) MRI changes in status epilepticus. Neurology 60: 1866.
-
Leach JP, Chadwick DW, Miles JB, Hart IK (1999) Improvement in adult-onset Rasmussen's encephalitis with long-term immunomodulatory therapy. Neurology 52: 738-742.
-
Takahashi Y, Yamazaki E, Mine J, Kubota Y, Imai K, et al. (2013) Immunomodulatory therapy versus surgery for Rasmussen syndrome in early childhood. Brain Dev 35: 778-785.
-
Bien CG, Tiemeier H, Sassen R, Kuczaty S, Urbach H, et al. (2013) Rasmussen encephalitis: Incidence and course under randomized therapy with tacrolimus or intravenous immunoglobulins. Epilepsia 54: 543-550.
