

Clinical Medical
Reviews and Case Reports
Spondyloepiphyseal Dysplasia Tarda with Progressive Arthropathy Associated with Osteoporosis and Cataract: A Case was Misdiagnosed as Juvenile Idiopathic Arthritis
Fatma Gul Yurdakul*, Rezan Kocak, Filiz Sivas, Aysegul Altun Güvenir and Hatice Bodur
Ankara Numune Training and Research Hospital Department of Physical Medicine and Rehabilitation, Turkey
*Corresponding author:
Fatma Gul Yurdakul, Ankara Numune Training and Research Hospital Department of Physical Medicine and Rehabilitation Talatpasa Boulevard No: 5 Altindag-Ankara, Turkey, Tel: 05059254214, E-mail: fatmagulonder@gmail.com
Clin Med Rev Case Rep, CMRCR-2-062, (Volume 2, Issue 10), Case Report; ISSN: 2378-3656
Received: August 07, 2015 | Accepted: October 06, 2015 | Published: October 09, 2015
Citation: Yurdakul FG, Kocak R, Sivas F, Güvenir AA, Bodur H (2015) Spondyloepiphyseal Dysplasia Tarda with Progressive Arthropathy Associated with Osteoporosis and Cataract: A Case was Misdiagnosed as Juvenile Idiopathic Arthritis. Clin Med Rev Case Rep 2:062. 10.23937/2378-3656/1410062
Copyright: © 2015 Yurdakul FG, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Spondyloepiphyseal dysplasia tarda with progressive arthropathy (SEDT-PA) is rare hereditary disorder with autosomal recessive, X-linked recessive or autosomal dominant inheritance. Because of joint involvement SEDT-PA may be confused with juvenile idiopathic arthritis and SEDT-PA also called progressive pseudorheumatoidarthropathy of childhood. The present case had been misdiagnosed as juvenile arthritis and received unnecessary treatments for years.
A 21-year-old male patient admitted with swelling and deformity in the fingers of hands, walking disturbance and common joint paint. Anteroposterior X-ray of the hip joint revealed widened and flattened epiphyses of the femoral head and bilateral narrow and irregular joint spaces. Bilateral tibiofemoral and patellofemoral joint spaces were narrowed in knee radiograph and there was osseous enlargement of the tibiofemoral joints. X-ray of the hands showed periarticular osteoporosis, significant narrowing of the joint spaces of proximal interphalengial and distal interphalangeal joints and osseous enlargement of the basis of the metacarpal, proximal and distal phalengeal bones. In lateral vertebral radiograph there was generalized osteopenia, platyspondyly with loss of weight in vertebral bodies, increased kyphosis in lateral thoracic spine and increased lordosis in lateral lumbal spine. He was diagnosed as SEDT-PA with osteoporosis and cortical cataract. Clinical features and joint involvement of SEDT-PA are similar to the JIA. Early diagnosis of SEDT-PA recovers patients from inappropriate and unnecessary treatments. Osteoporosis and ocular anomalies should be considered in patients diagnosed with SEDT-PA.
Keywords
Spondyloepiphyseal dysplasia, Juvenile arthritis, Osteoporosis, Cataract, Misdiagnose
Introduction
The skeletal dysplasias (osteochondrodysplasias) are a group of rare disorders that characterized by common abnormalities in skeleton. The incidence of these disorders is 1/5000. Radiographic abnormalities were evaluated to classify skeletal disorders. In the radiographic classification, these disorders are named according to the pathological area (epiphyseal, metaphyseal, diaphyseal) of the bone. Disorders are named as spondyloepiphyseal dysplasia (SED) when the pathology is in the vertebral epiphysis [1]. SED is defined as 3 clinic groups: 1- SED congenita 2-SED tarda 3-SED tarda with progressive arthropathy (SEDT-PA) [2]. The skeletal dysplasias are genetically heterogeneous and SEDT usually inherited as X-linked recessive. It is characterized by short trunk and extremities, deformity of the vertebraes, platyspondyly, kyphoscoliosis, coxa vara, genu valgum- varum, abnormal shape and structure of the epiphyses of hand bones. Boys are affected more often [1-3]. Although the one set of SEDT- PA is 3 and 6 years of age, they are diagnosed at an older age. Because of joint involvement SEDT-PA may be confused with juvenile idiopathic arthritis and SEDT-PA also called progressive pseudo rheumatoidarthropathy of childhood [4-6].
Case Report
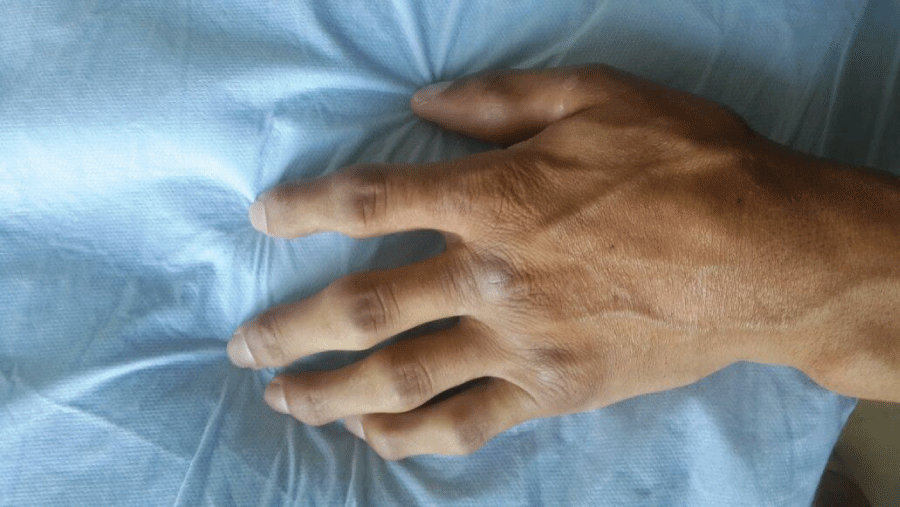
A 21-year-old male patient who lives in rural area admitted to outpatient clinic of physical medicine and rehabilitation with swelling and deformity in the fingers of hands, walking disturbance and common joint paint. It started first with low back pain, pain in the hip, knee and ankle joints since 6 years of age. Pain and swelling started in his wrists and metacarpophalangeal (MCP) and proximal interphalengial (PIP) joins about 10 years old. He had and stiffness lasting for 2 hours. He had been diagnosed as juvenile arthritis. Past medical records were not received. He had received various analgesics and non steroidal anti-inflammatory drugs from different physicians, but he did not remember steroid and DMARD (disease-modifying anti-rheumatic drugs) use. His complaints did not respond to these medical treatments. He had no fever, skin rash, uveitis and muscle weakness. His neuromotor development was normal.
There was consanguineous marriage of his parents. His family members including parents, 4 brothers and 3 sisters had no similar symptoms. His intelligence was normal and had uncomplicated birth history.
Physical examination: His height was 151 cm (vertex to pubis 70 cm and pubis to floor 81 cm), arms span was 166 cm, and weight was 43 kg. Thoracic kyphosis and lumbar lordosis had increased slightly. His skin, neurological, cardiorespiratory and genitourinary system examinations was normal. He had normal audiometric results and his visual acuity was 0.4 and 1.0 in the right and left eyes. There was bilaterally cortical cataract.
The abduction and flexion of shoulders, the extension of elbows and knees, abduction and internal- external rotation of hips are limited. His wrists and ankles are ankylosed in neutral position. Interphalaengial joints of fingers swollen with flexion deformity. His cervical and lumbar spine extensions were restricted with pain (Figure 1).
.
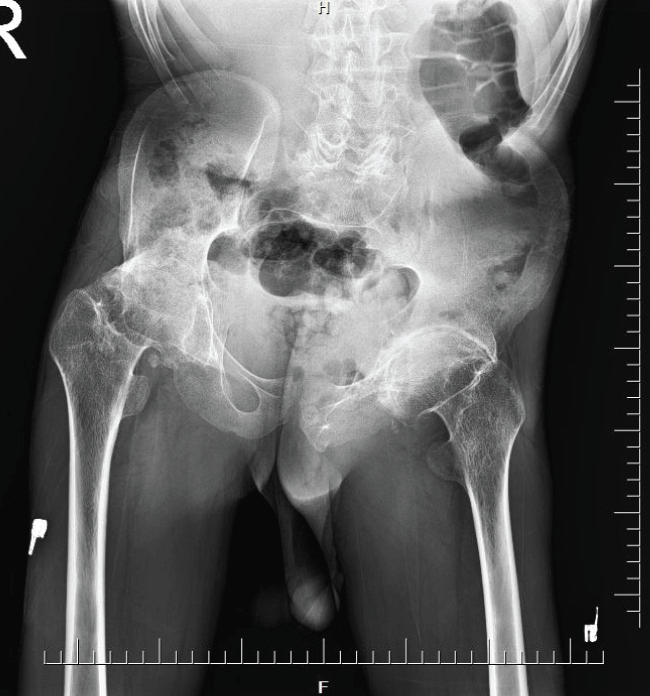
Figure 2: Widening of the epiphyses of femoral heads and changes in coxofemoral joints.
View Figure 2
In laboratory analysis complete blood count, liver and kidney function, electrolytes, proteins, lipids and routine urinalysis were normal. The erythrocyte sedimentation rate was 11 mm/h, C reactive protein was 2 mg/L (0.2-5 mg/L), and anti-streptolysin-O < 200 Todd U. Rheumatoid factor, anticycliccitrullinated peptide, antinuclear antibody, hepatitis markers, Rose Bengal agglutination, celiac antibodies were negative. Levels of anti-DNA, complement C3 and C4 were normal, the thyroid stimulating hormone, testosterone, thyroid function tests, parathyroid hormone levels, and protein electrophoresis were normal. Vitamin D level was 15.38 ng/ml (30-100 ng/ml).
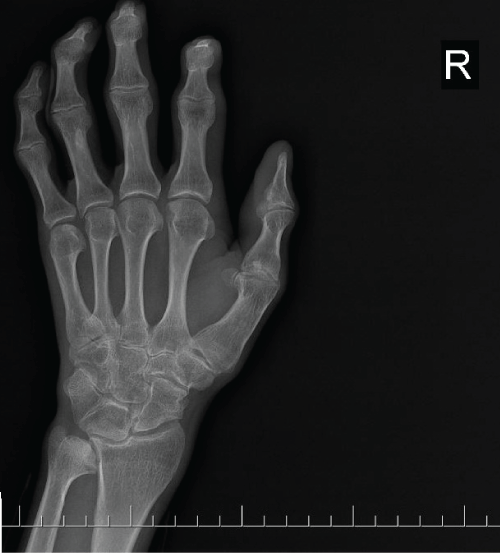
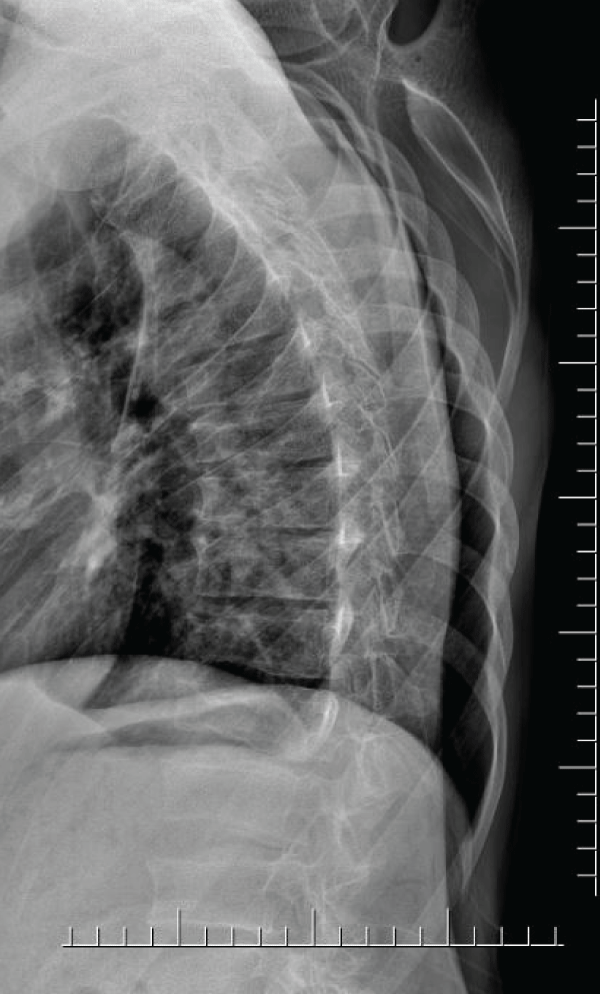
Anteroposterior X-ray of the hip joint revealed widened and flattened epiphyses of the femoral head and bilateral narrow and irregular joint spaces (Figure 2). Bilateral tibiofemoral and patellofemoral joint spaces were narrowed in knee radiograph and there was osseous enlargement of the tibiofemoral joints. X-ray of the hands showed periarticular osteoporosis, significant narrowing of the joint spaces of PIP and distal interphalangeal (DIP) joints and osseous enlargement of the basis of the metacarpal, proximal and distal phalengeal bones (Figure 3). In lateral vertebral radiograph there was generalized osteopenia, platyspondyly with loss of weight in vertebral bodies, increased kyphosis in lateral thoracic spine and increased lordosis in lateral lumbal spine (Figure 4).
.
Figure 4: Generalized osteopenia, platyspondyly with loss of weight in vertebral bodies, and increased kyphosis in lateral thoracic spine.
View Figure 4
Because of generalize osteopenia in vertebral radiographs osteoporosis was assessed with dual energy X-ray absorptiometry (DEXA). L2-L4 bone mineral density was 0.759 g/cm2, T-score was -3.0, Femur total bone mineral density was 0.718 g/cm2, T-score was -2.4.
He was diagnosed as SEDT-PA with osteoporosis and cortical cataract. He received vitamin D supplementation for Vitamin D deficiency and continued with vitamin D-calcium preparations and antiresorptive treatment. He was advised physiotherapy of joints, informed about his the disease and exercise programs.
Discussion
SEDT-PA is a rare hereditary disorder with autosomal recessive, X- linked recessive or autosomal dominant inheritance [1-3,7]. It is characterized by irregularities of the end plates of vertebral bodies, swelling and deformities of small and large peripheral joints [6,8]. According to the revision of the International Nomenclature and Classification of the Osteochondrodysplasias SEDTA-PA was listed in the group of "other spondyloepi-(meta)-physealdysplasias" [9]. SEDT-PA should be differentiated from juvenile idiopathic arthritis (JIA) to avoid the patients from unnecessary drug use and to ensure the optimal treatment [10]. The absence of laboratory changes indicating systemic or synovial inflammation and lack of response to anti rheumatic drugs help the differential diagnosis of SEDT-PA from JIA [8,11].
To the best of our knowledge the first case with SEDT-PA was described in 1982. Although the most common initial symptoms of SEDT are low back pain, difficulty in walking and pain in the hips and knees, the disease rarely starts with small peripheral joint involvement. In SEDT patients the age of one set changes between 3-11 years old however the reported age at diagnoses ranges from 4-58 years old [4-20]. The diagnosis is delayed due to the slow progression of symptoms, the rarity of the disease and JIA like joint involvement. Initial symptoms, age of one set and clinical features of some cases in literature are located in table 1 [4-8,10-20]. Our patient had low back pain, pain in hips and knees since 6 years old. Swelling and pain in hand joints started in 10 years old. These features are similar to most of cases we have seen in the literature.
![]()
Table 1: Characteristics of patients diagnosed with SEDT or SEDT-PA, presented in the literature.
View Table 1
Short stature and longer arm span are known features of the SEDT. Patients' stature changes between 132- 165 cm and arm spans are 152-170 cm [4-7,11,19,20]. The present case height is 151 cm and arm span is 166 cm. He is not taller than the other patients described in the literature. His arm span is longer than his height and upper segment (vertex to pubis) is shorter than lower (pubis to floor) like other patients.
Spine complaints of our patients started in 6 years old and involvement of peripheral joint started in 10 years old. Because of the short stature, affected thoracic and lumbar spine, and hand joints involvement mimicking JIA he was considered as SEDTA-PA. Although clinical findings are similar to JIA, there was no evidence of inflammation in the laboratory findings and radiographic findings were consistent with SEDT-PA. Autosomal dominant and X-linked inheritance was not considered due to the lack of similar complaints from his patients, sisters and brothers. It is compatible with autosomal recessive inheritance in this patient.
SEDT cases have been published in the literature and disorders have been reported accompanying the SEDT-PA. Reported disorders include corneal changes and osteoporosis [6,16,19,20]. Although the generalized osteopenia can be seen with SEDT, publications indicate the presence of osteoporosis is limited [13,15,18]. Osteoporosis in SEDT has been reported in 2 cases and both of these cases lumbar L2-4 T score were lower than the hip T score [19,20]. Our patient's lumbar L2-4 t score was -3.0 and it was lower than hip T score. In addition our patient had also vitamin D deficiency. There were no malabsoption and hydroxylation disorders in this patient. He received insufficient sunlight due to the joint pain and walk disorder. Physician should keep in mind that patients with SEDT-PA may have osteoporosis and it should be noted that while the treatment.
Another disorder accompanied by SEDT is about cornea and lens. Corneal opacities reported in a patient with SEDT and subcapsuler cataract in a patient with SEDT-PA [6,16]. SEDT-PA is known as a form of type II collagenopathy and defect in collagen type II may lead to ocular anomalies [16]. In our patient there was bilaterally cortical cataract. The reduced visual acuity in right eye was considered about amblyopia. Patients with SEDT-PA might have vision problems at an early age and a multidisciplinary approach with ophthalmologist is important.
The treatment of these patients is symptomatic. Growth hormone or anabolic steroid therapy for treatment of short stature was considered but the results were found contradictory. It has been reported in that anabolic steroid therapy can be useful in the treatment of short stature in the patients with skeletal dysplasia in early childhood [21]. On the other hand Burrens' study results showed that growth hormone therapy is not effective for treatment of short stature in skeletal dysplasia [22]. Specific stretching and strengthening exercises with encouragement for active life are useful therapies for the patients with skeletal dysplasia. Arthroplasty is known to be an option in cases with persistent pain [11]. The present case was advised physiotherapy (Transcutaneous Electrical Nerve Stimulation) for joint pains, informed about his the disease and exercise programs.
In conclusion SEDT-PA is a rare hereditary arthropathy. Clinical features and joint involvement are similar to the JIA. Early diagnosis of SEDT-PA recovers patients from inappropriate and unnecessary treatments. Osteoporosis and ocular anomalies should be considered in patients diagnosed with SEDT-PA.
References
-
O'Dell JR, Firestein GS, Budd RC, Gabriel SE, McInnes IB, et al. (2013) Heritable Disease of Connective Tissue. In: Kelley's Textbook of Rheumatology. Philadelphia: Elsevier Saunders 1719-1737.
-
Lateur ML, Klippel JH, Dieppe PA (1994) Bone and Joint Dysplasias. In: Rheumatology. London: Mosby-Year Book Europe Limited 7: 1-10.
-
Mc Alister WH, Resnick D, Niwayama G (1988) Osteochondrodysplasias and other Skeletal Dysplasias -Diagnosis of Bone and Joint Disorders. Philadelphian 3442-3515.
-
Sarioglu S, Arasil T, Kucukdeveci A, Gursel Y (2003) Spondyloepiphesael Dysplasia Tarda: A Case Report. T Klin J PM&R 3: 93-96.
-
Arslanoglu S, HizarciogluM, Genel F (2000) Spondyloepiphyseal dysplasia tarda with progressive arthropathy:An important form of osteodysplasia in the differential diagnosis of juvenile rheumatoid arthritis. Pediatrics International 42: 561-563.
-
Mandal SK, Ghosh S, Mondal SS, Chatterjee S (2014) Spondyloepiphyseal dysplasia tarda with progressive arthropathy associated with subcapsular cataract. BMJ Case Rep 2014.
-
Tug E, Senocak E (2008) Spondyloepiphyseal Dysplasia Tarda with Progressive Arthropathy with Delayed Diagnosis. Turk J Med Sci 38: 83-89.
-
Bal S, Kocyigit H, Turan Y, Gurgan A, Bayram KB, et al. (2009) Spondyloepiphyseal dysplasia tarda: four cases from two families. Rheumatol Int 29: 699-702.
-
Lachman RS (1998) International nomenclature and classification of the osteochondrodysplasias (1997). Pediatr Radiol 28: 737-744.
-
Bennani L, Amine B, Ichchou L, Lazrak N, Hajjaj-Hassouni N (2007) Progressive pseudorheumatoid dysplasia: three cases in one family. Joint Bone Spine 74: 393-395.
-
Kocyigit H, Arkun R, Ozkinay F, Cogulu O, Hizli N, et al. (2000) Spondyloepiphyseal dysplasia tarda with progressive arthropathy. Clin Rheumatol 19: 238-241.
-
Wynne-Davies R, Hall C, Ansell BM (1982) Spondylo-epiphysial dysplasia tarda with progressive arthropathy. A "new" disorder of autosomal recessive inheritance. J Bone Joint Surg Br 64: 442-445.
-
Rasore-Quartino A, Camera A, Camera G (1993) Spondylo-epiphyseal dysplasia tarda with progressive arthropathy: description of a patient whose mother showed minimal features of the disease. Pathologica 85: 225-231.
-
el-Shanti HE, Omari HZ, Qubain HI (1997) Progressive pseudorheumatoid dysplasia: report of a family and review. J Med Genet 34: 559-563.
-
Cogulu O, Ozkinay F, Ozkinay C, Sapmaz G, Yalman O, et al. (1999) Progressive pseudorheumatoid arthropathy of childhood. Indian J Pediatr 66: 455-460.
-
Hirata Y, Watanabe H, Maeda N, Inoue Y, Shimomura Y, et al. (2000) Corneal changes in spondyloepiphyseal dysplasia tarda. Jpn J Ophthalmol 44: 29-32.
-
Ehl S, Uhl M, Berner R, Bonafe L, Superti-Furga A, et al. (2004) Clinical, radiographic, and genetic diagnosis of progressive pseudorheumatoid dysplasia in a patient with severe polyarthropathy. Rheumatol Int 24: 53-56.
-
Kaptanoaylu E, Perain F, Perain S (2004) Spondyloepiphyseal dysplasia tarda with progressive arthropathy. Turk J Pediatr 46: 380-383.
-
Kurtulmus S, Bayram KB, Kocyigit H, Koksal-Avci S, Dincer-TuranY, et al. (2006) Spondyloepiphyseal Dysplasia Tarda and Osteoporosis: A Case Report. Osteoporoz Dunyasindan 12: 18-21.
-
Batmaz I, Sariyildiz MA, Dilek B, Ulu MA, VerimS, et al. (2013) A Case of Spondyloepiphyseal Dysplasia Tarda Coexisting With Osteoporosis and Mimicking Spondyloarthropathy. Turk J Phys Med Rehab 59: 260-263.
-
Buyukgebiz A, Kovanlikaya I (1993) Oxandrolone therapy in skeletal dysplasia. Turk J Pediatr 35: 189-196.
-
Burren CP, Werther GA (1996) Skeletal dysplasias: response to growth hormone therapy. J Pediatr Endocrinol Metab 9: 31-40.
