

Obstetrics and Gynaecology Cases - Reviews
An Uncommon Case of Antepartum and Postpartum Surgical Aorta Repair in a Patient with Marfan Syndrome
Liggins Casandra A*, Lin Monique G and Foley Michael R
Department of Obstetrics and Gynecology, Banner Good Samaritan Medical Center, USA
*Corresponding author: Casandra A. Liggins, MD, Department of Obstetrics and Gynecology, Banner Good Samaritan Medical Center, 1111 East McDowell Rd, Phoenix, AZ 85045, USA, E-mail: Casandra.liggins@bannerhealth.com
Obstet Gynecol Cases Rev, OGCR-1-014, (Volume 1, Issue 2), Case Report; ISSN: 2377-9004
Received: November 09, 2014 | Accepted: November 28, 2014 | Published: November 30, 2014
Citation: Liggins CA, Lin MG, Foley MR (2014) An Uncommon Case of Antepartum and Postpartum Surgical Aorta Repair in a Patient with Marfan Syndrome. Gynecol Cases Rev 1:014. 10.23937/2377-9004/1410014
Copyright: © 2014 Liggins CA, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Background: Marfan syndrome is a heritable connective tissue disorder with significant risk of cardiovascular compromise. The physiological changes of pregnancy greatly increases that risk resulting in significant threat of maternal and fetal morbidity and mortality.
Case: A 21-year-old G1P0 female with Marfan syndrome presented at 16 weeks gestation with a 7.3 cm ascending aortic aneurysm and a desire to maintain her pregnancy. Immediate surgical repair was performed upon presentation with reassuring maternal and fetal status until cesarean section delivery at 39 weeks gestation. The patient�s postpartum course was complicated by an expanding descending aortic aneurysm which, upon advancement to a type B aortic dissection, required additional surgical management.
Conclusion: The risk of cardiovascular sequelae associated with Marfan syndrome in pregnancy is significant though it can be lessened with preconception counseling and potential surgical intervention. In the event of cardiovascular compromise during pregnancy, endovascular or surgical repair versus medical management are treatment options dependent on patient presentation, acuity and available resources.
Keywords
Marfan syndrome, Aortic dissection, Aorta graft, Pregnancy
Introduction
Marfan syndrome is a rare, heritable disease that results in altered connective tissue structure. Persons with Marfan syndrome are at risk for cardiovascular compromise, most significantly; aortic dissection. The risk of aortic dissection increases significantly during pregnancy, primarily due to the physiological changes pregnancy induces. There is significant morbidity and mortality associated with Marfan syndrome in pregnancy, during both the intrapartum and postpartum period. Although principles of management remain the same, the additional life at risk in pregnancy complicates the treatment approach. We present a case of rapid and acute ascending aortic dilation in a Marfan patient during the second trimester with subsequent descending aortic dissection following delivery.
Case
A 21-year-old, G1P0 female with Marfan syndrome presented to the hospital at approximately 16 weeks gestational age for surgical management of a 7.3 cm thoracic aortic aneurysm. The patient had been seen by her obstetrician for acute onset of chest pain and was found to have a murmur on examination. She was subsequently evaluated by a cardiologist where an echocardiogram revealed a fusiform aneurysm extending from the aortic root to the distal ascending aorta, measuring 7.3 cm in cross-section at the mid ascending aorta with no evidence of dissection (Figure 1). The great vessels of the arch, celiac, SMA, renal and IMA were widely patent. In addition, left ventricular enlargement was seen with an ejection fraction of 45-50% (Figure 2). At time of admit, the patient was counseled extensively about her options including proceeding with the pregnancy while on beta blocker management, though this option was not encouraged, termination of the pregnancy and proceeding with surgical repair of her aneurysm or undergoing surgical repair of her aorta while attempting to maintain the pregnancy. Despite the risk of maternal and fetal compromise, the patient opted for surgical repair while maintaining the pregnancy. Aortic valve replacement with a 25 mm Edwards pericardial tissue valve, replacement of the ascending aorta with a 28 mm sinus of Valsalva tube graft and reimplantation of the right and left coronary arteries was performed. The patient was placed on cardiopulmonary bypass during the repair with antegrade cold blood cardioplegia which was given in retrograde fashion throughout the case. Her ejection fraction at conclusion of the case was 30-35%. Following the surgery, an ultrasound of the fetus was performed demonstrating live intrauterine fetal gestation with a heart rate of 148 bpm. The patient�s postoperative course was significant only for a left sided pleural effusion for which she underwent thoracentesis without complication. She was discharged on labetalol six days following the procedure.
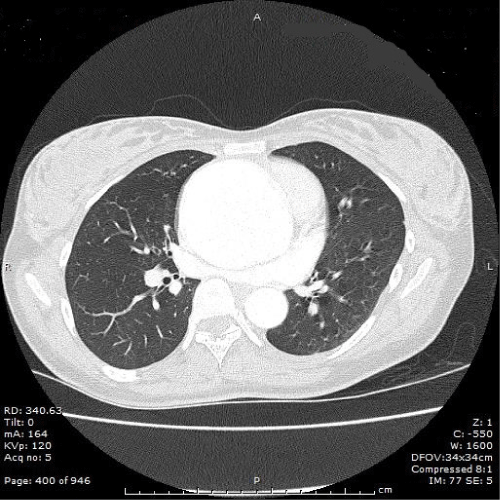 Figure 1: Computed tomography angiogram chest/abdomen/pelvis with
contrast at 16 weeks gestation. Fusiform aneurysm from the aortic root
to the distal ascending aorta, maximum 7.3cm in the mid ascending. No
evidence of dissection.
View Figure 1
Figure 1: Computed tomography angiogram chest/abdomen/pelvis with
contrast at 16 weeks gestation. Fusiform aneurysm from the aortic root
to the distal ascending aorta, maximum 7.3cm in the mid ascending. No
evidence of dissection.
View Figure 1
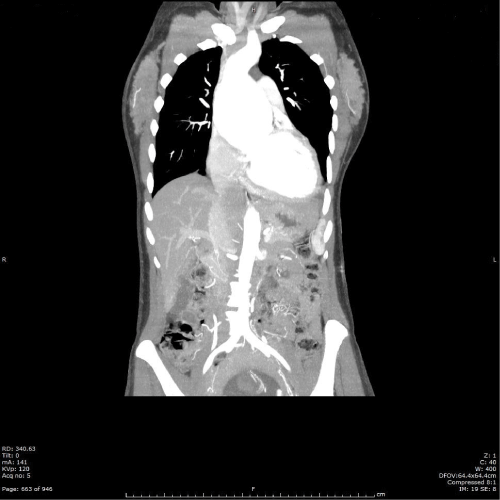 Figure 2: Computed tomography angiogram chest/abdomen/pelvis with
contrast at 16 weeks gestation. Enlarged left ventricle suggesting volume
overload aortic regurgitation.
View Figure 2
Figure 2: Computed tomography angiogram chest/abdomen/pelvis with
contrast at 16 weeks gestation. Enlarged left ventricle suggesting volume
overload aortic regurgitation.
View Figure 2
The patient was monitored by chest computed tomography approximately 3 months later and was found to have no evidence of ascending aortic aneurysm following the graft placement. However, a 37 mm proximal descending aortic aneurysm was identified, which had expanded from 30 mm on imaging three months prior. She continued beta blocker therapy and the remainder of her pregnancy was uneventful. She was admitted to the hospital at 39 weeks, 3 days gestational age for elective primary cesarean section delivery. As the patient had remained on beta blocker therapy through the duration of her pregnancy, fetal growth was assessed at the time of delivery. The fetus had demonstrated appropriate growth with an estimated fetal weight of 3224 grams, at the 43%ile. At time of admission, extensive counseling was provided to the patient about subsequent pregnancies. She declined bilateral tubal ligation at time of cesarean section and voiced a desire to conceive again in the future. Cesarean section was performed without complication and with reassuring neonatal status having Apgar�s of 9 and 9 at 1 and 5 minutes, respectively. On postoperative day 2, the patient complained of new onset and worsening chest pain. Chest computed tomography revealed an enlarging fusiform descending thoracic aortic aneurysm of 41mm in addition to an enlarging abdominal aorta extending from the level of the celiac artery measuring 22mm to below the renal arteries measuring 18 mm, both had increased 2 mm from prior imaging. Aggressive blood pressure control was initiated to maintain systolic blood pressure below 100 bpm in an attempt to stabilize the aortic expansion. Repeat chest computed tomography imaging the following day showed degeneration of the descending aortic aneurysm to a type B aortic dissection (Figure 3) and although the patient was maintained on oral antihypertensives, her blood pressures remained labile. As such, she was taken back to the operating room 8 days following her cesarean delivery for surgical management of the aneurysmal degeneration of a type B aortic dissection. Replacement of the descending aorta with a 20 mm Hemashield tube graft and repair of the right common femoral artery and vein was performed while under cardiopulmonary bypass with deep hypothermic circulatory arrest. Intraoperatively, the patient was found to have a large entry tear just distal to the subclavian artery and an exit tear just before the diaphragm. Following repair, the patient returned to the intensive care unit for close monitoring and support. Later that evening, the patient was found to have a large left chest wall hematoma. She returned to the operative room for exploration and evacuation of the hematoma. She again returned to the intensive care unit and her remaining postoperative course was uneventful. She was discharged home on hospital day 17.
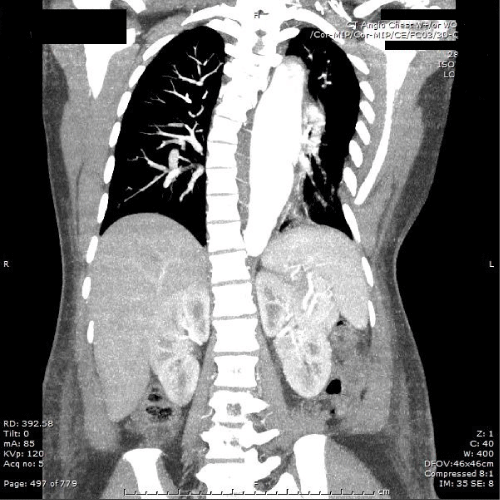 Figure 3: Computed tomography angiogram chest with contrast in
postpartum. New descending thoracic aortic dissection and interval
enlargement of the fusiform descending thoracic aneurysm from one day
prior, now maximum 45mm.
View Figure 3
Figure 3: Computed tomography angiogram chest with contrast in
postpartum. New descending thoracic aortic dissection and interval
enlargement of the fusiform descending thoracic aneurysm from one day
prior, now maximum 45mm.
View Figure 3
Following discharge, the patient was readmitted approximately 1 month later. She was found to have a left groin seroma and acute-on-chronic systolic heart failure with an ejection fraction that decreased from 35 to 15%. She had been noncompliant with outpatient diuretics which were restarted, resulting in improvement in her volume status over the course of 6 days. She was discharged with a LifeVest and close outpatient cardiology follow-up. Unfortunately, the patient remained noncompliant and approximately 2 weeks later she was found unresponsive at home. Upon arrival of emergency medical services, she was in v-fib cardiac arrest. She was resuscitated and placed on the hypothermia protocol. She experienced two additional cardiac arrests due to torsades de pointes while in the intensive care unit. At this point, the patient remains hospitalized with non-ischemic cardiomyopathy and an ejection fraction of 10-15%. Although she has survived her pregnancy and enjoys her newborn, she faces a lifetime of the cardiovascular repercussions that pregnancy and noncompliance has afforded her.
Discussion
Marfan syndrome is an autosomal dominant connective tissue disorder caused by a FBN-1 gene mutation [1]. The syndrome has a high penetrance with variable expression of cardinal features comprising cardiovascular, ocular and skeletal manifestations. Persons with Marfan syndrome are predisposed to thoracic aortic aneurysms and dissections as well as mitral valve prolapse and regurgitation. Aortic dissections occur from a disruption of the media layer of the aorta and are often resultant from an aortic aneurysm [2]. According to the Stanford classification, type A dissection involves the ascending aorta with or without involvement of the descending aorta, whereas a type B dissection involves the descending aorta alone [3,4]. Aortic dissection in pregnancy and those associated with Marfan syndrome are most often type A [3,5]. Specifically, it is approximated that 65% of tears occur in the ascending aorta, 30% occur in the descending aorta, less than 10% occur in the aortic arch and approximately 1% occur in the abdominal aorta [6]. Surgical repair is recommended for type A dissections and typically involves endovascular stents or grafts to restore the true vessel lumen and to reestablish distal perfusion [2,3,6]. Classically, the treatment of uncomplicated type B dissections is with medical management involving beta blockade aimed at decreasing the heart rate and reducing sheer stress on the vessel [2,6]. Complicated type B dissections involving rupture, end organ ischemia, aneurysmal aortic expansion, extension of the dissection or persistent pain, require surgical intervention [3,4].
The incidence of acute aortic dissection in pregnancy is low, 0.4 per 100,000, approximately 60-80% of which are thoracic in location with the proximal tear occurring within the ascending aorta [1,7]. Of the remaining type B dissections, half will occur in the postpartum period [3]. Marfan syndrome has been reported in up to 50% of aortic dissections occurring in pregnancy and survival rates have ranged from 20-30% with a drastic 1% per hour incremental maternal mortality [6]. Physiologic adaptations of pregnancy including increased blood volume, heart rate, stroke volume and cardiac output in addition to increased aortic compliance in the setting of decreased tissue elasticity in Marfan syndrome, confers the additional risk of aortic dissection during pregnancy. Pregnancy, as an independent risk factor, induces degenerative alterations in the aortic media through a shift in hormone levels that persist beyond delivery [3]. Rising levels of estrogen and progesterone hormones and receptors in the wall of the aorta results in loss of vessel-wall integrity. Most aorta pathology occurs in the third trimester as that is the point of maximal hemodynamic stress. These alterations increase with advanced gestational age and parity, as does the risk of maternal morbidity and mortality [3,8].
Women with Marfan syndrome experience an increased risk of aortic dissection, even in the absence of preconception cardiovascular abnormality [4]. However, the risk increases substantially in patients with aortic diameters greater than 4 cm and those that are associated with aortic valve disease or rapid aortic dilation [7,9]. For women with an aortic diameter greater than 4.0 cm, prophylactic surgery prior to conception is recommended as more than half will undergo surgery during pregnancy, experience a rupture or will have life-threatening growth during pregnancy [2]. For those women who undergo repair during pregnancy, fetal loss during hypothermia and cardiopulmonary bypass is common as is the risk of severe hypoxic-ischemic insult [2,8]. However, avoiding hypothermia precludes open repair of the aorta. Thus, the use of antegrade cerebral perfusion for brain protection and moderate hypothermia can be used, as described in the reported case [8]. In the event of stable aortic dilation or type B dissection, continued management in pregnancy with the use of beta blockers is a reasonable alternative. Due to the risk of reduced fetal growth often seen with beta blocker administration, frequent monitoring should be performed during the pregnancy [2]. Overall, the risk of maternal and/or fetal morbidity and mortality including intrauterine fetal growth restriction, preterm delivery, cesarean section and operative vaginal delivery is higher in those with Marfan syndrome [2,5,10].
Optimal management during pregnancy of a patient with Marfan syndrome includes preconception counseling involving prophylactic aortic grafting if the aortic diameter exceeds 4.0cm [8]. Frequent echocardiogram monitoring throughout pregnancy should be used to identify acute changes in cardiovascular status and inpatient management should be initiated at 28-32 weeks gestation. If the aortic root is greater than 4.0 cm and stable or type B dissection is observed in the antepartum, beta blocker therapy should be initiated and continued for 3 months postpartum [8]. Surgical intervention in the event of rapid aortic dilation or dissection should occur upon diagnosis, regardless of dissection type, as the delay of intervention is associated with significant maternal and fetal mortality[6]. Dissections discovered in the third trimester should be surgically managed at time of cesarean delivery [11]. In the event of a type B dissection, beta blockers can be used during pregnancy to reduce cardiac and vascular strain with additional close monitoring of fetal growth. However, endovascular repair is necessary if the type B dissection is unstable or complicated [3]. Additionally, patients should be informed of the increasing risk of fetal and/or maternal demise with each subsequent pregnancy. Adequate preconception counseling is necessary to significantly reduce the risk to mother and baby, whether the patient is nulliparous or multiparous.
References
-
Donnelly RT, Pinto NM, Kocolas I, Yetman AT (2012) The immediate and long-term impact of pregnancy on aortic growth rate and mortality in women with Marfan syndrome. J Am Coll Cardiol 60: 224-229.
-
Hiratzka LF, Bakris GL, Beckman JA, Bersin RM, Carr VF, et al. (2010) 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM Guidelines for the diagnosis and management of patients with thoracic aortic disease: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. Circulation 121: e266�e369.
-
Rosenberger LH, Adams JD, Kern JA, Tracci MC, Angle JF, et al. (2012) Complicated postpartum type B aortic dissection and endovascular repair. Obstet Gynecol 119: 480-483.
-
Braverman AC (2010) Acute aortic dissection: clinician update. Circulation 122: 184-188.
-
Curry RA, Gelson E, Swan L, Dob D, Babu-Narayan SV, et al. (2014) Marfan syndrome and pregnancy: maternal and neonatal outcomes. BJOG 121: 610-617.
-
Yuan SM (2013) Aortic dissection during pregnancy: a difficult clinical scenario. Clin Cardiol 36: 576-584.
-
Kim SW, Kim D, Hong JM (2014) Acute aortic dissection in pregnancy with the marfan syndrome. Korean J Thorac Cardiovasc Surg 47: 291-293.
-
Immer FF, Bansi AG, Immer-Bansi AS, McDougall J, Zehr KJ, et al. (2003) Aortic dissection in pregnancy: analysis of risk factors and outcome. Ann Thorac Surg 76: 309-314.
-
Lipscomb KJ, Smith JC, Clarke B, Donnai P, Harris R (1997) Outcome of pregnancy in women with Marfan's syndrome. Br J Obstet Gynaecol 104: 201-206.
-
Hassan N, Patenaude V2, Oddy L2, Abenhaim HA1 (2014) Pregnancy Outcomes in Marfan Syndrome: A Retrospective Cohort Study. Am J Perinatol .
-
Brautbar A, LeMaire SA, Franco LM, Coselli JS, Milewicz DM, et al. (2010) FBN1 mutations in patients with descending thoracic aortic dissections. Am J Med Genet A 152A: 413-416.
