

Obstetrics and Gynaecology Cases - Reviews
Termination of Pregnancy for an 11q Terminal Deletion Incidentally Diagnosed Prenatally; Jacobsen Syndrome Penetrance and Ethical Dilemmas
Sofia F Makrydima*, Eftihios Trakakis, Athanasios Minkoff and Nikolaos Papantoniou
3rd Department of Obstetrics and Gynaecology, Attikon University Hospital, Athens, Greece
*Corresponding author: Sofia F Makrydima, 3rd Department of Obstetrics and Gynaecology, Attikon University Hospital, Rimini 1, 12462, Haidari, Athens, Greece, Tel: 00306976198815/00447477855193, E-mail: soma20med@hotmail.com
Obstet Gynecol Cases Rev, OGCR-3-092, (Volume 3, Issue 4), Case Report; ISSN: 2377-9004
Received: January 29, 2016 | Accepted: June 24, 2016 | Published: July 05, 2016
Citation: Makrydima SF, Trakakis E, Minkoff A, Papantoniou N (2016) Termination of Pregnancy for an 11q Terminal Deletion Incidentally Diagnosed Prenatally; Jacobsen Syndrome Penetrance and Ethical Dilemmas. Obstet Gynecol Cases Rev 3:092. 10.23937/2377-9004/1410092
Copyright: © 2016 Makrydima SF, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
The currently noted boost of de novo microdeletions has been partly attributed to the relevant increase of mean paternal age at the time of first child acquisition. The simultaneous widespread implementation of molecular techniques for prenatal karyotyping has revealed distinct deletion aberrations of uncertain clinical significance. Recent research has failed to confirm the intrinsic correlation between implicated genome fragment and extent of phenotypic abnormality. We present a case of an 11q terminal deletion detected incidentally in a fetus displaying no evidence of anomaly during successive detailed ultrasound anatomy scans. The genotype corresponds to Jacobsen syndrome exhibiting great heterogeneity attributed to epigenetic modifications. For example, homeostatic mechanisms at the level of transcription or translation can mitigate the phenotypic effects of a deletion. However, unmasking of a recessive mutation on the non- deleted homologous chromosome can amplify phenotypic abnormalities. Genetic counselling is a priori considered as a challenging process taking into consideration that the spectrum of normal is subjectively defined by the personal perspectives of the individual. Undoubtedly, it becomes even more demanding if no tangible deformity can be depicted. Reporting bias lurks when genetic consultation is mainly based on previously recorded phenotypes, since less affected persons are unlikely to be detected.
Keywords
Jacobsen syndrome, 11q Terminal deletion, Genetic counseling, Phenotypic variability, Antenatal diagnosis
Introduction
Before the advent of molecular techniques, prenatal diagnosis of chromosomal abnormalities was based, exclusively, on conventional cytogenetic analysis. Cells were arrested midmitosis and chromosomes were analyzed microscopically after the addition of a specific banding agent. During the past decade, microdeletions have been increasingly detected due to the widespread use of newer molecular methods, including microarray comparative genomic hybridization (a CGH) and single nucleotide polymorphism genotyping arrays (SNP) [1].
As a result, a growing number of non- lethal syndromes have been established and attributed to microdeletions. It is remarkable that the majority of these syndromes share common phenotypic characteristics including short stature and distinct dysmorphic facial features such as dolichocephaly or trigonocephaly, hypertelorism, epicanthal folds, downslanted eyes or palpebral fissures, broad nose bridge, short upturned nose, low set ears, thin upper lip, palatal defects, receding chin (micrognathia) and short webbed neck. More importantly, they are associated with congenital defects, involving cardiac or renal anomalies, which usually can be depicted at the second trimester ultrasound scan. The impacted degree of cognitive impairment remains the most controversial feature, taking into consideration that its' extent cannot be predicted antenatally, neither can it be appreciated immediately postnatally [2].
By definition, microdeletion syndromes have been identified due to the corresponding clinical phenotype. Their genetic basis has only been investigated in the aftermath. Prenatal counselling entails a reversal of the aforementioned process, thus, clinical geneticists are requested to foresee the clinical phenotype of an individual based on molecular karyotyping.
The "genotype-first" approach can be very challenging given the possibility of genotype- phenotype discordance. Variable expressivity can result from various breakpoints; copy number variations or single nucleotide polymorphisms in non- coding regions. The cornerstone of phenotype heterogeneity is reduced (or incomplete) penetrance, an emerging phenomenon that has precipitated our understanding of Mendelian inheritance. Oligogenic inheritance, genomic imprinting (parent of origin effect), allele dosage, modifier genes and gene- environmental interplay may account for the severe inconsistencies in clinical presentation of people carrying the same mutations. Velocardiofacial/ di George syndrome is a characteristic example of reduced penetrance; significant clinical differences have been reported even among monozygotic twins, suggesting non- genetic modifications [3].
The presented case refers to a prenatal diagnosis of a terminal 11q deletion following elective amniocentesis. The lack of tangible deformities at the ultrasound scan coupled with restrictions in predicting phenotypic variability, at both the genetic and epigenetic level, raised severe ethical dilemmas.
The Case
Α 43-year-old woman, married to a 57-year-old man for seven years, visited the outpatient's department of our clinic claiming infertility and chronic pelvic pain. The patient had a history of a first trimester spontaneous abortion (gravida 1, para 0) with the same husband. Ultrasound examination revealed endometriosic cysts and the patient underwent a diagnostic laparoscopy. During the procedure, the cysts were removed and the peritoneal cavity was carefully inspected for infertility related findings. There were no significant adhesions, whereas dye test confirmed tubal patency. There was no uterine anomaly identified. The woman was discharged the day following the procedure.
Immediately after the operation, the patient conceived, but she remained unaware of her pregnancy until she reached 20 weeks. The second trimester scan could not detect any major or minor anomaly, but for a single umbilical artery. The patient opted for invasive prenatal testing and she underwent amniocentesis on the 22nd week of pregnancy. After three weeks, a large terminal deletion disorder was found 46, XY, del (11) (q23q25) using conventional karyotyping. Undoubtedly, fluorescence in situ hybridization or microarray comparative genomic hybridization would allow more reliable genotype- phenotype correlations. However, further investigation was not feasible because of financial restrictions.
Genetic counselling
The specific chromosomal perturbation has been closely linked to Jacobsen syndrome (OMIM: 147791), which carries an ambiguous prognosis, with signs and symptoms that vary considerably among affected individuals. Psychomotor retardation constitutes the most prevalent disorder, while behavioral impairment has commonly been reported, including compulsive behavior, attention deficit hyperactivity disorder (ADHD) and autism spectrum disorders. The syndrome is also characterized by the distinct facial stigmata described previously for all microdeletion syndromes. More than 90% of patients suffer from a rare bleeding disorder called Paris- Trousseau syndrome, involving thrombocytopenia, pancytopenia and platelet function disorders. Other clinical manifestations can encompass congenital heart defects, skeletal/ limb abnormalities, pyloric stenosis, recurrent infectious episodes and genitourinary anomalies [4].
The information delivered to the woman had a detrimental effect on her psychology, causing an intense grief reaction that culminated to suicidal ideation. A team of mental health professionals, who examined her, diagnosed anxiety disorder. As outlined, giving birth to a child suffering from Jacobsen syndrome, as presented in literature, could add severe psychological burden to her. In addition, the parents postulated that they couldn't support a child with learning difficulties that needs early intervention. Besides, the country suffers a deficient social care system, access to benefits and occupational therapists is limited, whereas public education regarding living with disability is atrocious.
In view of the responsibility and the diligence assigned to bringing up a child with disability, the couple finally decided to terminate the pregnancy. They proceeded to feticide, followed by induction of labor, since the fetus had reached viability. During the procedure a detailed scan depicted normal anatomy (Figure 1), which was further attested by the obstetrician and the midwife who examined the dead neonate immediately after birth. It is worth mentioning that the parents refused postmortem pathology analysis of the fetus.
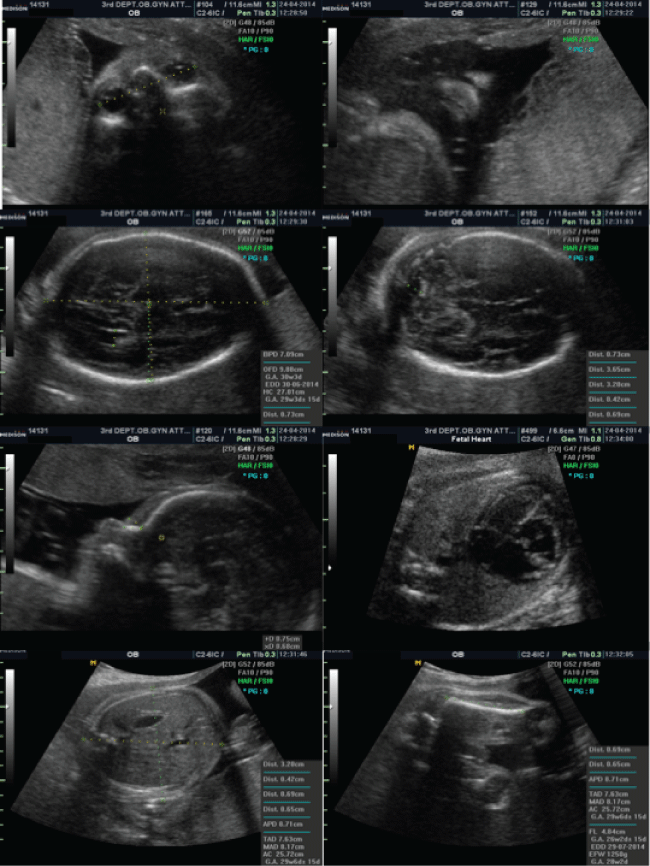
.
Figure 1: Detailed scan of the fetus with the 11q terminal deletion performed just before the feticide depicted normal anatomy (29th week of gestation).
View Figure 1
Prenatal diagnosis of Jacobsen syndrome has been reported in the literature, but to our knowledge this is the first case where no structural disorder could be identified in the second trimester scan, not even in the detailed scan performed at the time of feticide [5,6].
Informed consent
Informed consent was obtained from the participant couple for being included in the report. All procedures followed were in accordance with national guidelines and the ethical standards on research involving human subjects as defined by the World's Medical Association Declaration of Helsinki, last revised in 2013.
Discussion/Interpretation
Pathophysiology of deletion mutations
Jacobsen syndrome results from de novo mutations in 85-90% of cases or can be inherited when a parent carries a balanced translocation in 10-15% of cases. Chromosome 11 is involved in the most common reciprocal translocation observed in humans, t (11; 22) (q23; q11). Unfortunately, the parents did not proceed to self- karyotyping, as advised, mainly because they could not afford the cost. In addition, they selected to abandon the idea of children acquisition, influenced by this emotionally traumatic event and their advanced age. The possibility that the deletion was the unbalanced product of a familial reciprocal translocation cannot be excluded. The fact that the woman reported at least one known early abortion reinforces this assumption.
Paternal age can also be linked to the deletion, especially if it was a de novo one. In a landmark paper, Kong et al. presented data supporting that paternal age constitutes the dominant factor in determining the number of de novo mutations in the child. The effect is linear and it accounts for an increase of about two mutations per year. The finding is in consistency with the physiology of spermiogenesis, which means that the observed higher germline mutation rate in males can be attributed to the higher number of germ cell divisions [7]. Taking into account relative trials, the authors raised the possibility that the recently recorded trend in autism diagnosis is at least partially due to an increase in the average age of father at the time of conception.
Phenotypic variability
Traditional karyotyping remains the standard of care for prenatal diagnosis and reveals large aberrations of genetic material, which are considered a priori, deleterious. Depending on the extent of the aneuploidy, the gene expression system is distorted and the fitness of the organism challenged. Intuitively, scientists hypothesized that the larger the fragment involved, the more possible to result in phenotypic disease. But this concept is extremely simple- minded and recent advances in genetics have shed light to epigenetic homeostatic mechanisms trying to re-balance genomic irregularities. The phenomenon known as "dosage compensation" at the level of transcription or translation may counteract, to some extent, for addition or loss of chromosomal segments. At least three compensatory responses to aneuploidies have been described: buffering, feedback and feed forward.
It has been shown that only 29% of the expressed chromosome 21 transcripts (either genes or open reading frames) are over expressed in Down syndrome and account for the observed phenotype. The other 71% of expressed sequences are either compensated (56%) or highly variable among individuals (15%) [8]. Interestingly, buffering can be more effective in case of deficiencies than duplications, by increasing the steady-state mRNA levels originating from the remaining allele [9].
A review of the literature confirms that the severity of the observed clinical abnormalities in patients with JS is not clearly correlated with the extent of the deletion [10]. Indeed, intellectual disability results from complex gene interactions. Despite the fact that critical gene domains have been identified, a firm conclusion cannot be drawn. Recently, researchers failed to show a statistically significant relationship between cognitive-behavioral characteristics or developmental trajectories and deletion size [11]. Surprisingly, a small terminal deletion of 2,2 Mb of the 11q chromosome has been described in a two-year-old boy showing no evidence of the syndrome. The deleted fragment is known to contain 21 genes, seven of which have OMIM annotations [12]. Nevertheless, some characteristics that display high penetrance can be exclusively affiliated with specific genes, such as platelet abnormality mapping to FLI-1 gene 11q24 [13].
Whenever a deletion occurs, any recessive mutation on the non- deleted homologous chromosome would become unmasked. Obviously, such events would be rare and they may be responsible for uncommon features observed in a minority of affected individuals. For example, they could provide explanation for case reports such as a patient with cleft palate in the context of JS [14]. It is reasonable, that this mechanism could operate in more subtle ways. It could unmask DNA- sequence polymorphisms on the intact chromosome that affect the expression of genes associated with cognitive function or neuropsychiatric profile [15].
Review of the Literature
An incidental diagnosis of Jacobsen syndrome has never been published in the international literature before. Valduga et al. described a mosaic 46,XY [16]/46,XY,del(11)(q23), identified on fetal karyotype because of a serum screening test positive for Down syndrome. Mosaicism rate was estimated at 20% and neither the successive anatomy ultrasound scans, performed till the 31st week of pregnancy, nor the postmortem examination suggested a remarkable abnormality [16]. Parental consultation under these circumstances can be very intriguing, since there is no definite detectable anomaly. Current practice entails mapping the genotype using phenotypic characteristics. Consequently, clinical geneticists can present the notions derived from the 200 cases of Jacobsen syndrome recorded worldwide and research using genetically engineered animal models to depict causal genes.
However, scientific community cannot exclude the possibility that an equal number of similar genotypic individuals manifesting milder phenotypes may exist, but have not been identified because of the lack of distinct characteristics. Publication bias, implicating selection and reporting of the worst-case scenarios, lurks and may result in misleading the parents. Researchers' innate belief that deletions always cause some sort of functional incompetence, incompatible with self service and social integration, remains to be proved. Undoubtedly, up to date there are no data regarding the number of people that carry the same chromosomal aberration and what percentage of them experience significant disorders or disabilities.
Evidently, these ethical dilemmas are further amplified when it comes to mutations not visible at the level of conventional karyotype. The application of molecular karyotyping has revealed numerous microdeletions, which are very difficult to interpret, in particular when they are de novo and they cause no obvious anatomic abnormality. As a result there has been a growing concern that expanding prenatal screening could cause trivialization of abortions and ultimately lead to the "Gatacca" era [17,18]. Researchers have drawn attention to such ethical issues; Chervenak and McCullough propose that non- lethal anomalies on a viable fetus, should drive directive recommendation against aggressive management for the sake of maternal benefit [19].
Research Recommendations
To adequately understand the molecular effects of chromosomal aberrations and their role in disease etiology, there is need of large-scale genomic approaches. Whole genome sequencing of general population together with simultaneous transcriptome analysis could pinpoint silent mutations and estimate the component of gene expression determined by an individual's genetic profile. Trends are shifting towards whole-genome tissue-dependent prediction models, such as PrediXcan, that are trained with reference transcriptome data sets [20]. It is highly possible that in the near future prenatal screening will provide meaningful options for reproductive choice to pregnant women and their partners.
Conclusions
The increase of mean paternal age at first conception, which is noted at modern society, has led to a surge of distinct chromosomal deletions. The molecular techniques implemented as part of prenatal screening have contributed to the detection of such aberrations. Although, there has been considerable progress to mapping human genome and explaining epigenetic adjustments, the phenotype of the individual cannot be definitely predicted. Therefore, clinical geneticists may not be ready to handle the results of molecular karyotyping as part of antenatal testing in a low risk population, where no significant anatomic abnormality can be depicted during the typical ultrasound scans. Besides, genetic counselling can be tricky by definition, since the spectrum of "normality" is arbitrary determined according to the individual's beliefs.
Acknowledgments
We would like to thank Dr. Athena Soukafor performing the ultrasound scans and for her contribution to the management of this case.
Declaration of Interest
Author Makrydima, author Trakakis, author Minkoff and author Papantoniou report no declaration of interest.
References
-
Kloosterman WP, Hochstenbach R (2014) Deciphering the pathogenic consequences of chromosomal aberrations in human genetic disease. Mol Cytogenet 7: 100.
-
Vissers LE, Stankiewicz P (2012) Microdeletion and microduplication syndromes. Methods Mol Biol 838: 29-75.
-
Carvill GL, Mefford HC (2013) Microdeletion syndromes. Curr Opin Genet Dev 23: 232-239.
-
Mattina T, Perrotta CS, Grossfeld P (2009) Jacobsen syndrome. Orphanet J Rare Dis 4: 9.
-
Chen CP, Chern SR, Chang TY, Tzen CY, Lee CC, et al. (2004) Prenatal diagnosis of the distal 11q deletion and review of the literature. Prenat Diagn 24: 130-136.
-
Boehm D, Laccone F, Burfeind P, Herold S, Schubert C, et al. (2006) Prenatal diagnosis of a large de novo terminal deletion of chromosome 11q. Prenat Diagn 26: 286-290.
-
Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, et al. (2012) Rate of de novo mutations and the importance of father's age to disease risk. Nature 488: 471-475.
-
Ait Yahya-Graison E, Aubert J, Dauphinot L, Rivals I, Prieur M, et al. (2007) Classification of human chromosome 21 gene-expression variations in Down syndrome: impact on disease phenotypes. Am J Hum Genet 81: 475-491.
-
Prestel M, Feller C, Becker PB (2010) Dosage compensation and the global re-balancing of aneuploid genomes. Genome Biol 11: 216.
-
Nalbantoglu B, Donma MM, Nişli K, Paketçi C, Karasu E, et al. (2013) Jacobsen syndrome without thrombocytopenia: a case report and review of the literature. Turk J Pediatr 55: 203-206.
-
Fisch GS (2015) Cognitive-behavioral characteristics and developmental trajectories in children with deletion 11qter (Jacobsen syndrome), and their relation to deletion size. Am J Med Genet A 167: 45-53.
-
Evers C, Janssen JW, Jauch A, Bonin M, Moog U (2012) A small terminal deletion 11q in a boy without Jacobsen syndrome: narrowing the critical region for the 11q Jacobsen syndrome phenotype. Am J Med Genet A 158: 680-684.
-
Favier R, Akshoomoff N, Mattson S, Grossfeld P (2015) Jacobsen syndrome: Advances in our knowledge of phenotype and genotype. Am J Med Genet C Semin Med Genet 169: 239-250.
-
Ysunza A, Shaheen K, Aughton DJ, Micale MA, Merson R, et al. (2013) Velopharyngeal insufficiency, submucous cleft palate and a phonological disorder as the associated clinical features which led to the diagnosis of Jacobsen syndrome. Case report and review of the literature. Int J Pediatr Otorhinolaryngol 77: 1601-1605.
-
Grossfeld PD, Mattina T, Lai Z, Favier R, Jones KL, et al. (2004) The 11q terminal deletion disorder: a prospective study of 110 cases. Am J Med Genet A 129: 51-61.
-
Valduga M, Cannard VL, Philippe C, Romana S, Miton A, et al. (2007) Prenatal diagnosis of mosaicism for 11q terminal deletion. Eur J Med Genet 50: 475-481.
-
de Jong A, de Wert GM (2015) Prenatal screening: an ethical agenda for the near future. Bioethics 29: 46-55.
-
Fischer S (2015) State of the ART: Emerging genetic technologies in reproductive medicine are rapidly making real what once was science fiction? are we ready for it? IEEE Pulse 6: 8-13.
-
Chervenak FA, McCullough LB (1996) The fetus as a patient: an essential ethical concept for maternal-fetal medicine. J Matern Fetal Med 5: 115-119.
-
Gamazon ER, Wheeler HE, Shah KP, Mozaffari SV, Aquino-Michaels K, et al. (2015) A gene-based association method for mapping traits using reference transcriptome data. Nat Genet 47: 1091-1098.
