

Journal of Genetics and Genome Research
Identification of Ten Novel Mutations in Factor VIII Gene: A Study of A Cohort of 52 Haemophilia A Patients
Rosa Santacroce 1*,Angelica Leccese 1, Roberta Trunzo1, Giuseppe Lassandro2, Paola Giordano2, Cosimo Ettorre3, Stefano Antoncecchi4, Isabella Cantori5, Alfredo Dragani6, Donata Belvini7, Roberta Salviato7, and Maurizio Margaglione1
1Department of Clinical and Experimental Medicine, University of Foggia, Italy
2Department of Biomedical Science and Human Oncology, University of Bari "Aldo Moro", Italy
3Hemophilia and Thrombosis Center, Policlinico Giovanni XXIII, Italy
4Transfusion Medicine, Haemophilia Centre San Giacomo Hospital, Italy
5Haemophilia Center, Civil Hospital, Italy
6Bleeding and Thrombotic Disorders Unit, Department of Hematology, Ospedale Civile dello Spirito Santo, Italy
7Transfusion Service, Hemophilia and Regional Blood Disease Centre, Castelfranco Veneto Hospital, ULSS 8 Regione Veneto, Italy
*Corresponding author: Rosa Santacroce, Department of Clinical and Experimental Medicine, University of Foggia, Via Luigi Pinto 1, 71122 Foggia, Italy, Tel: +39 0881 733842; Fax: +39 0881 736082; E-mail: rosa.santacroce@unifg.it
J Genet Genome Res, JGGR-1-006, (Volume 1, Issue 1), Research Article; ISSN: 2378-3648
Received: September 24, 2014 | Accepted: October 10, 2014 | Published: October 14, 2014
Citation: Santacroce R, Leccese A, Trunzo R, Lassandro G, Giordano P, et al. (2014) Identification of Ten Novel Mutations in Factor VIII Gene: A Study of A Cohort of 52 Haemophilia A Patients. J Genet Genome Res 1:006. 10.23937/2378-3648/1410006
Copyright: © 2014 Santacroce R, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Introduction: Haemophilia A (HA) is the most common X-linked recessive genetic disease caused by mutations in the gene coding for coagulation factor VIII (FVIII) resulting in spontaneous bleeding.
Aim: The aim of our study is to provide additional information about the genetic causes of HA describing the correlation between the observed mutations and the clinical phenotype in a cohort of 52 patients suffering from HA to different degrees.
Methods: First we performed a search of inversion 22 (IVS22) and of the intron 1 (IVS1) for severe HA patients; inversions negative and moderate/mild HA patients were screened by direct sequencing of the coding regions and exon-intron junctions of FVIII gene. Where no PCR amplification was observed we used the Multiplex Ligation-dependent Probe Amplification (MLPA) and, to confirm a large deletion, we performed an array-comparative Genomic Hybridization (array CGH).
Results: 42% of severe HA patients had IVS22 while the remainder had missense mutations, large deletions and small insertions. All but one moderate/mild HA patients had missense mutations. Only two patients did not show good correlation between the genotype and the clinical phenotype reported to us by the respective centers of haemophilia. We also identified 10 novel mutations not previously described in the most common mutation database.
Conclusion: We studied a group of 52 HA patients and we found 26 mutations in the FVIII gene of which 10 are new mutations. These results confirm the great heterogeneity of the molecular defects gene responsible for the deficiency of FVIII.
Keywords
Haemophilia A, FVIII gene, Molecular analysis, Genotype-Phenotype correlation, Novel mutations, Mutation database
Introduction
Haemophilia A (HA) is a recessive inherited X-linked blood coagulation disorder, due to mutations in procoagulant factor VIII (FVIII) that affects 1 in 6000 males. The FVIII gene maps on the distal end of the long arm of the X-chromosome (Xq28), spans 186 kb and comprises 26 exons [1]. HA is usually diagnosed during the first year of life in affected males although there are reports of female carriers with mild clinical manifestations of the HA disease. The severity of the disease is determined by activity levels of circulating factor VIII (FVIII:C) allowing a classification into 3 forms: severe (FVIII:C< 1% of normal), moderate (FVIII:C 1-5% of normal) and mild (FVIII:C >5% - < 40% of normal) HA disease [2]. In the HAMSTeRS factor VIII mutation database [3] are reported 2107 unique mutations, the majority of which (938) are missense mutations which, as is clear from the literature data, are reported in 37% of severe, 72% of moderate and 88% of mild haemophilia A patients [4]. The FVIII intron 22 inversion is the causative mutation in 45% of severe HA patients [5] while FVIII intron 1 inversion has a frequency of 1.8% [6] in severe HA population. The remaining patients negative for intron 22/1 inversion and missense mutations present small deletions, nonsense mutations, splice-site mutations, small insertions, large deletions and others mutations involving different mechanisms [4].
We investigated FVIII gene abnormalities in 52 haemophilia A patients from different areas of Italy and from other countries, and we found 26 mutations, 16 of which are already described in the HAMSTeRS database and/or in the Human Gene Mutations Database (URL: http://www.hgmd.org) and/or CHAMP F8 Mutation List (http://www.cdc.gov/ncbddd/hemophilia/champs.html) that compared to the previous two database considers additional publications; the other 10 mutations are not reported in these database.
Material and Methods
Informed consent was obtained from the patients after approval of the local Human Ethics Committees. The studies were carried out according to the Principles of the Declaration of Helsinki.
Patients and DNA sample
We collected blood samples, 5ml in sodium citrate, from 52 HA patients, from different Italian haemophilia treatment centers (Macerata, Bari, Pescara and Foggia). These 52 patients comprised 26 (50%) severe, 3 (5,8%) moderate and 23 (44,2%) mild cases based on FVIII:C.
Genomic DNA was purified from 200 ml of whole blood sample type using QIAamp DNA Blood Mini Kit (QIAGEN®).
Molecular analysis
The molecular analysis of FVIII gene was carried out depending on the severity of haemophilia A as this influences the diagnostic strategy used [7]. First of all we screened severe HA patients for the FVIII intron 22 (IVS22) and later for intron 1 (IVS1) inversion mutation. The IVS22 was analysed by Long PCR method [8] and IVS1 by a multiplex PCR [9]. Severe HA patients negative for inversion mutations together with moderate and mild HA patients were analysed for the exons and exon-intron junctions of the entire FVIII gene by direct sequencing using the BigDye® TerminatorTM method on an ABI PRISM 3100 Genetic Analyzer sequencer (Applied Biosystems, Foster City, California, USA) according to the manufacturer's instructions. The sequence data results from the analysis of sequencing were compared with the reference NM_000132 sequence of the FVIII gene deposited in NCBI database. The detected mutations, reported in accordance to the Human Genome Variation Society (HGVS http://www.hgvs.org/mutnomen/), were confirmed by repeating the amplification and sequencing reaction of interested FVIII gene fragments. Patients in which PCR amplification of FVIII gene exons failed were analysed by Multiplex Ligation-dependent Probe Amplification (MLPA) using SALSA MLPA probemix P178-B2 F8 Lot B2-0312 (MRC-Holland, Amsterdam, and the Netherlands). The reaction products were detected on an ABI PRISM 3130 Genetic Analyzer (Applied Biosystems, Foster City, California, USA). To assess the presence of large deletions, in some patients, an array-comparative Genomic Hybridization (array CGH) analysis was subsequently performed.
Bioinformatic analysis
For the interpretation of new mutations found, we used the Alamut® Visual software (available online at www.interactive-biosoftware.com/) that allows interpretation of variant pathogenicity. Alamut is a decision support application developed by Interactive Biosoftware, which is widely used in medical genetics that works with the aid of integrated functions such as: data integration from public data sources, conservation information, polymorphisms, published mutations and protein annotations.
Results
IVS22 and IVS1 mutations
11 (42%) of the 26 patients with severe haemophilia A had the IVS22 mutation while none had the IVS1 (Table 1). This results confirms the high percentage (45-50%) of IVS22 found in severe HA population [5]. Two severe HA brothers (FB19-FB20) showed a strange IVS22 band pattern with the only AB (10Kb) control band amplified using the Long PCR protocol. The mother of the two patients showed, however, the presence of the two bands AB and PQ of 10 and 12 Kb respectively. This result led to think of a possible deletion including the intragenic homologous copy int22h-1 amplified by the primer pair PQ.
![]()
Table 1:*Patients with de novo mutations (sporadic HA); Patients FB19 and FB20 are brothers, M2-M3 and C1-C2 are relatives; Patient P1 suffers from moderate haemophilia A (FVIII:C 2%) with presence of inhibitors. Mutations in bold are new mutations not described in HGMD, HAMSTeRS and CHAMP database; for those underlined have already been reported other mutations at the same site.
View Table 1
Missense and nonsense mutations
We found 15 missense mutations (Table 1). Of these five are novel mutations: c.943G>C (p.A315P), c.341C>A (p.P114H), c.1631A>G (p.D544G), c.530A>C (p.Y177S), c.6304G>A (p.G2102S); the nomenclature used in this work meets the HGVS instructions starting the nucleotide numbering from the A of the ATG start codon. Among four nonsense mutations two are not listed in the available database: c.4128C>G (p.Y1376X) and c.6045G>A (p.W2015X).
Small insertions and duplications
We also identified two novel insertions. The first one is the c.277_281insCAGGT (p.Pro93GlnfsX70), in exon 3, in a sporadic case of severe HA (FB2). The insertion of these five nucleotides leads to truncated protein due to the creation of a stop codon after 70 codons. The second one is an adenine duplication, c.3465dupA (p.Ser1156IlefsX10) in exon 14, which is not so far been reported in the aforementioned database and, like the previous one, create one stop site but only after 9 amino acids.
Deletions
A new deletion of 11 base pair c.5830_5840delATAATGGATAC was found in a severe HA patient. This caused a frameshift from codon 1944 in exon 18 with the consequent formation of a stop codon after 23 codons (p.Ile1944ThrfsX23). The other two deletions are described as large deletions detected by means of MLPA in five severe HA patients: g.EX1_EX22del [10] in FB19, FB20 (confirmed in their mother) and M23 patients. In FB19 and FB20 patients the array CGH revealed a 144 Kb deletion (arr[hg19] Xq28 (154,109,681-154,253,535)). The second deletion was g.EX10_EX11del [11] present in C1 and C2 patients.
Discussion
Haemophilia A is an X-linked disorder due to mutations in the FVIII gene, which is 186 kb long and consists of 26 exons. FVIII comprises a peptide leader of 19 amino acids and a mature protein of 2332 amino acids.
The present study aims to bring the spectrum of mutations that affect a heterogeneous population of patients with haemophilia A from various Italian haemophilia centers. We carried out the molecular analysis of the FVIII gene for all patients following a strategy of analysis depending on the clinical phenotype reported to us by the various haemophilia centers.
In our cohort of 52 HA patients we found 26 mutations including IVS22, missense mutations, nonsense mutations, large deletions, small insertions and small deletions. Of these 26 mutations 10 are novel mutations that have not been reported in the HAMSTeRS, HGMD and CHAMP database: five missense, two nonsense, two small insertions (one of which is duplication) and one small deletion (Table 1). The mutations not described were included in the online software Alamut® Visual to assess their pathogenicity (Figure1).
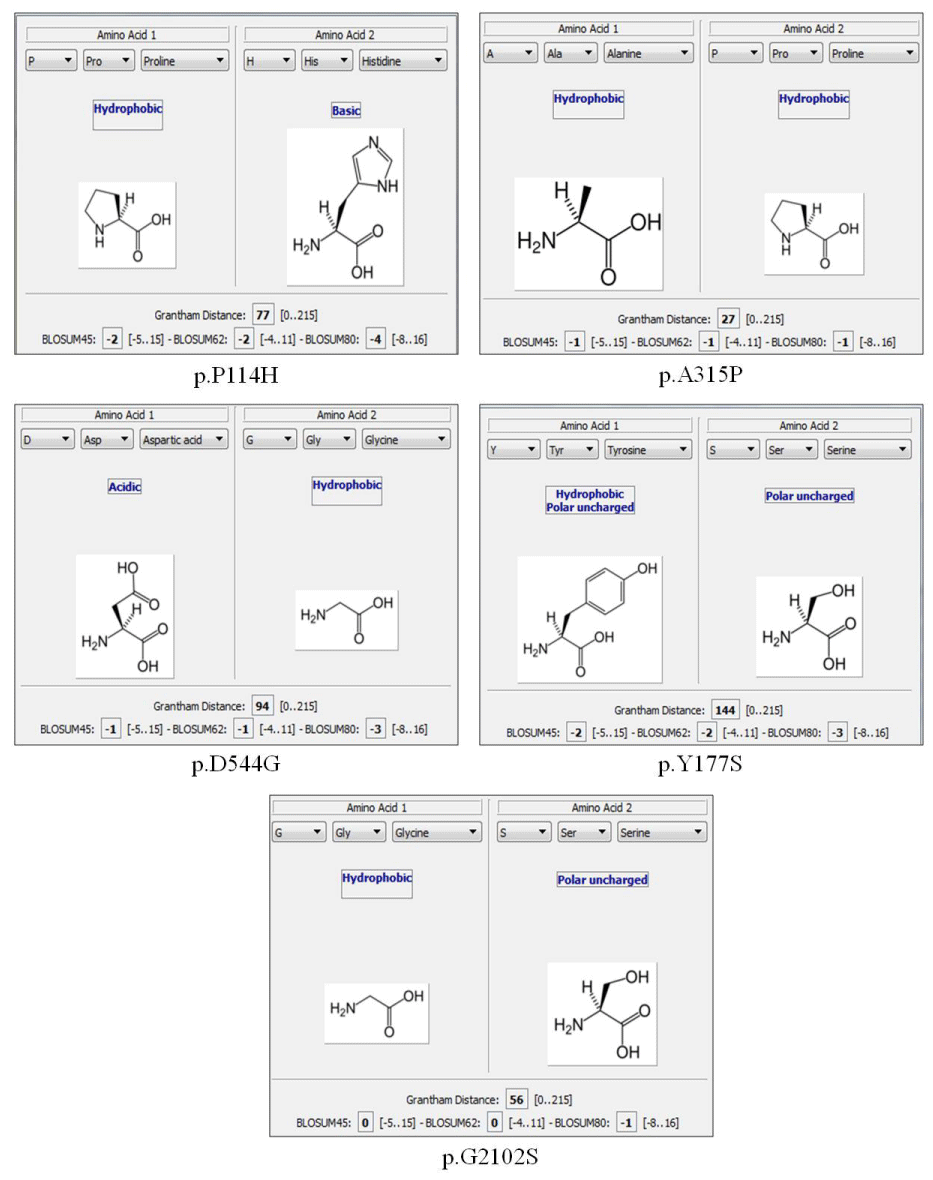
Figure 1: Results of Alamut software describing the functional changes of the five mutations not previously described
View Figure 1
Two of the novel mutations were found in patients (FB10-FB15) with sporadic severe haemophilia A.
In most cases the mutation found is able to explain the clinical phenotype of the patient, showing a good correlation between the genotype and HA phenotype. Our study presents cases (M4 and P1) that do not confirm this correlation.
The majority of frameshift mutations that create a premature stop codon have been reported in patients with severe hemophilia A. Despite this consideration in the literature are described such frameshift mutations, in exon 14 of FVIII gene, in moderate haemophilia A patients [12]. This is the case of patient M4, who suffers from mild haemophilia A, in which we found the mutation c.3465dupA, p.Ser1156fsX9 that creates a premature termination codon resulting in truncations in the B domain of the FVIII protein; thus the genotype and the clinical phenotype are discordant.
Patient P1 suffers from moderate haemophilia A with the presence of inhibitors; he showed c.5883G>A, p.W1961X mutation already reported in HAMSTeRS database but with different nucleotide transition (c.5882G>A) and only in severe HA patients and so the mutation found in patient P1 does not reflect his clinical phenotype.
The gross deletion g.EX1_EX22del was first found in FB19-FB20 brothers later the evidence of the anomalous INV22 band pattern. Subsequently MLPA assay was performed to assess the presence of an eventual deletion that has been confirmed by array CGH. Later, for patient M23 was used the same procedure for molecular diagnosis without making the array CGH.
In conclusion this study aims to validate the strategy of molecular analysis for the detection of mutations causative FVIII deficiency, confirming the great variability of this gene. We report 10 novel mutations spanning the entire FVIII gene that, in combination with all of that already given and those that will be identified, will benefit the genetic counseling and treatment of haemophilia A patients.
Acknowledgements
The authors stated that they had no interests which might be perceived as posing a conflict or bias.
Contributions of the authors:
i. R. Santacroce, A. Leccese performed the research.
ii. M. Margaglione designed the research study.
iii.R.Trunzo, G. Lassandro, P. Giordano, C. Ettorre, S. Antoncecchi, I. Cantori, A. Dragani, R. Salviato, D. Belvini contributed essential reagents or tools.
iv.R. Santacroce, A. Leccese wrote the paper.
References
-
Thompson AR (2003) Structure and function of the factor VIII gene and protein. Semin Thromb Hemost 29: 11-22.
-
White GC 2nd, Rosendaal F, Aledort LM, Lusher JM, Rothschild C, et al. (2001). Definitions in hemophilia. Recommendation of the scientific subcommittee on factor VIII and factor IX of the scientific and standardization committee of the International Society on Thrombosis and Haemostasis. Thromb Haemost 85: 560.
-
Kemball-Cook G. The Haemophilia A mutation, structure, test and resource site (HAMSTeRS). [http://hadb.org.uk/]
-
Santacroce R1, Acquila M, Belvini D, Castaldo G, Garagiola I, et al. (2008) The AICE-Genetics Study Group. Identification of 217 unreported mutations in the F8 gene in a group of 1,410 unselected Italian patients with hemophilia A. J Hum Genet (2008) 53:275-284.
-
Lakich D, Kazazian HH Jr, Antonarakis SE, Gitschier J (1993) Inversions disrupting the factor VIII gene are a common cause of severe haemophilia A. Nat Genet 5: 236-241.
-
Cumming AM; UK Haemophilia Centre Doctors' Organization Haemophilia Genetics Laboratory Network (2004) The factor VIII gene intron 1 inversion mutation: prevalence in severe hemophilia A patients in the UK. J Thromb Haemost 2: 205-206.
-
Keeney S, Mitchell M, Goodeve A on behalf of the UK Haemophilia Centre Doctors' Organisation (UKHCDO), the Haemophilia Genetics Laboratory Network and the Clinical Molecular Genetics Society (2010) Practice Guidelines for the Molecular Diagnosis of Haemophilia A.
-
Liu Q, Sommer SS (1998) Subcycling-PCR for multiplex long-distance amplification of regions with high and low GC content: application to the inversion hotspot in the factor VIII gene. Biotechniques 25: 1022-1028.
-
Bagnall RD, Waseem N, Green PM, Giannelli F (2002) Recurrent inversion breaking intron 1 of the factor VIII gene is a frequent cause of severe hemophilia A. Blood 99: 168-174.
-
Ma GC1, Chang SP, Chen M, Kuo SJ, Chang CS, et al. (2008) The spectrum of the factor 8 (F8) defects in Taiwanese patients with haemophilia A. Haemophilia 14: 787-795.
-
Rossetti LC, Radic CP, Candela M, Pérez Bianco R, de Tezanos Pinto M, et al. (2007) Sixteen novel hemophilia A causative mutations in the first Argentinian series of severe molecular defects. Haematologica 92: 842-845.
-
Akkarapatumwong V, Intorasoot S, Oranwiroon S, Thano-Otarakul P, Pung-Amritt P, et al. (2000) Frameshift mutations with severe and moderate clinical phenotypes in Thai hemophilia A patients. Hum Mutat 16: 530-531.
