

International Journal of Surgery Research and Practice
Congenital Cystic Lung Malformation Presenting in a Previously Asymptomatic Thirty-Year-Old Patient
Alecia Vandevelde*
Royal Darwin Hospital, Flinders University Darwin, Australia
*Corresponding author: Alecia Vandevelde MBBS/BA, Royal Darwin Hospital, 105 Rocklands Drive Tiwi, 0810 Northern Territory, Australia, E-mail: alecia.vandevelde@health.wa.gov.au
Int J Surg Res Pract, IJSRP-2-029, (Volume 2, Issue 2), Case Report; ISSN: 2378-3397
Received: September 04, 2015 | Accepted: October 23, 2015 | Published: October 26, 2015
Citation: Vandevelde A (2015) Congenital Cystic Lung Malformation Presenting in a Previously Asymptomatic Thirty-Year-Old Patient. Int J Surg Res Pract 2:029. 10.23937/2378-3397/1410029
Copyright: © 2015 Vandevelde A. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
This case is a rare presentation in an adult in his thirties of an uncommon disease normally detected in utero or shortly thereafter. It posed a diagnostic dilemma for our surgical and medical teams initially. We chose to manage this case surgically and have achieved a good outcome in doing so. The ideal management however is still controversial.
Keywords
Cystic lung disease, Congenital cystic adenomatoid malformation, Congenital cystic pulmonary malformation, Adults
Background
Congenital cystic adenomatoid malformations are usually detected antenatally or in early childhood. It is extremely rare for them to remain undetected and asymptomatic into adulthood. Ideal management in asymptomatic patients is controversial however due to the potential for malignant transformation excision is generally accepted as necessary.
Case Presentation
Our patient presented to the emergency department of our tertiary hospital with severe abdominal pain that had started suddenly. He was a fit thirty-year-old male who had no significant medical history apart from being an ex-smoker who was not on anymedications. He required significant amounts of opioid pain relief on arrival to control his pain.
The pain had initially come on two days preceding his presentation to the department however had settled. When it returned the next evening it was more severe, constant, andradiating from his left upper quadrant across to his epigastric region. He had not experienced the painpreviously. Although he had been undertaking moderately strenuous activity the day before the pain started he denied anypain during the activity and also denied any recent trauma. His bowels had been opening normally, he had no dysuria, and he did not usually suffer from symptoms suggestive of gastritis. He had not been drinking heavily preceding the presentation and had no history of gallstones nor did he have any symptoms suggestive of an upper respiratory tract infection. There was no recent unintentional weight loss or night sweats, and he had not been on any recent overseas travel.
On examination post significant amounts of morphine he appeared comfortable. He was afebrile and apart from a mild tachycardia he had otherwise unremarkable vital signs. His cardiovascular examination was normal, he had slightly decreased breath sounds at his left base and had mild flank tenderness. The remainder of his abdominal examination was unremarkable.
His urinalysis was normal and he had a white blood cell count of twenty, other routine bloods including urea and electrolytes, liver function tests, and lipase were unremarkable however. He went on to have an abdominal CT scan which extended to the lung bases and showed an unusual lesion sitting just above the diaphragm on the left side. Although at first glance similar in appearance to a diaphragmatic, the diaphragm appeared to be intact on the CT scan with no obvious extension of cystic contents below the diaphragm. The lesion was radio dense in appearance and sitting within the posterior basal left lower lobe measuring 94 by 56 mm in diameter. It had low density regions suggestive of fluid or necrosis. There was some associated atelectasis but no pleural effusion. There was no aberrant venous or arterial supply (Figure 1 and Figure 2).
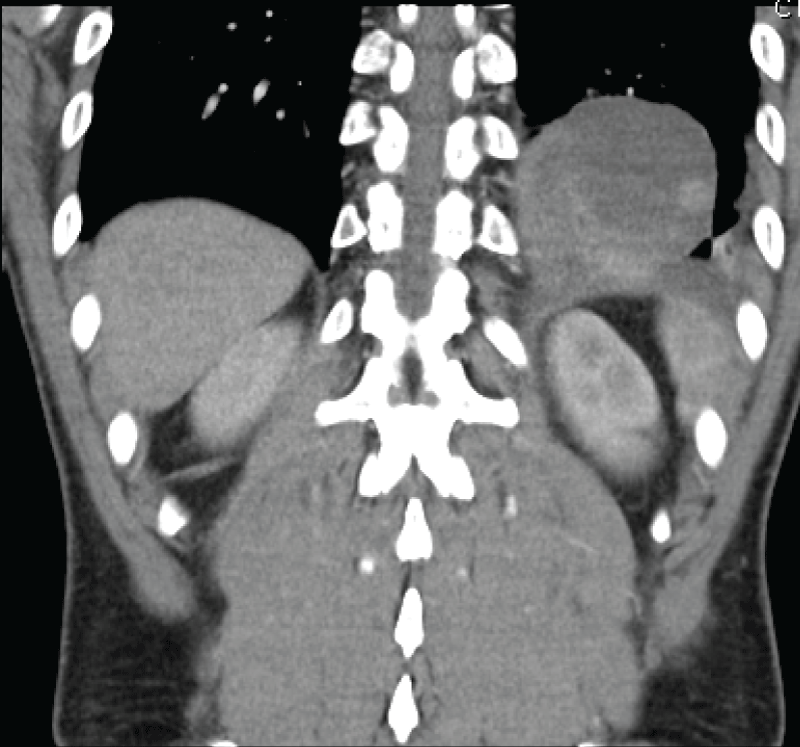
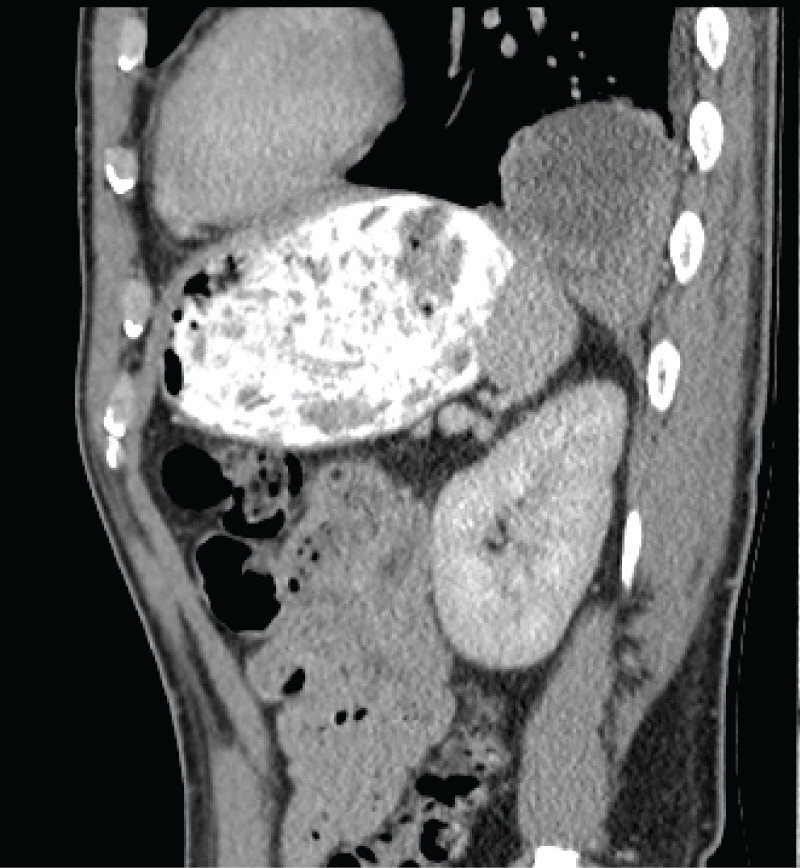
Further imaging with an ultrasound did not give a definitive diagnosis either. The scan revealed internal separations and variable wall thickness giving a differential of an empyema or a cystic mass.
MRI scan was not able to further determine the nature of the lesion either. It showed a mixed cystic and solid lesion probably lying within the pleural space rather than in the lung. The possible differential diagnoses postimaging included an infectious lesion, possibly a hydatid or empyema, a congenital lesion such aspulmonary sequestration, a neoplastic lesion was thought less likely but the comment that a sarcomacould not be ruled out was made. Open biopsy was suggested by the radiologists.
The patient went on to have a video assisted thorascopic excision biopsy of the lesion. The lesion was separate to the lung but attached with adhesions to the posterior tip of the left lung and so the lung was divided inferiorly with a GIA stapler leaving a portion attached to the cyst. Interestingly the lesion extended through a posterior defect in the diaphragm at the foramen of Bochdalek and a portion of the cyst lying quite close to the stomach was left in place to avoid perforation. An intraoperative gastroscopy was performed to ensure it was not in continuation with the stomach of which it was not. Grossly it was a cystic lesion measuring 70 × 35 × 30 mm containing mucous and old blood. Microscopically it was made up of cysts lined with ciliatedrespiratory epithelium. The walls revealed a benign cystic hamartoma with features consistent with atype I congenital cystic adenomatoid malformation. The walls of the cyst contained structuresresembling terminal bronchioles and some elastic fibres. Cartilage and clusters of seromucinous cells were found lining the spaces. There was no evidence of atypia, neoplasia, or malignancy. The cause of the patients acute pain leading to presentation is not entirely clear however could be potentially due to a bleed into the cyst. He recovered well post operatively and clinical follow up at one year has revealed no recurrenceof any respiratory symptoms or abdominal pain with the patient returning to full activity soon after the operation. He has not had any further imaging at this stage.
Discussion
With the advent of improved antenatal ultrasound screening congenital cystic adenomatoid malformations (CCAM) are generally detected in utero. Those that are not are most often picked up at a median age of 25 months. When symptomatic they present as respiratory distress in neonates and recurrent infections in older children [1]. The quoted incidence of CCLM varies from 1 in 10000 - 1 in 25000 depending on the study looked at. It is very uncommon for the lesions to remain undetected into adulthood, generally they are detected before the age of two [1,2]. CCAMs fall into the group of congenital cystic lung lesions along with bronchogenic cysts, congenital lobar emphysema, congenital parenchymal cysts, and pulmonary sequestrations [2,3]. When not diagnosed antenatally the lesions are often initially misdiagnosed as infections in children. In utero they can be asymptomatic or cause ascites, hydrothorax, fetal hydrops, and hypoplasia of the lung, situations occurring up to 25% of the time [3]. In a retrospective study done by Ramen looking at cases not detected in utero they found that x-ray detected the lesion only 45% of the time whereas CT scan detected the abnormality 100% of the time. Twenty percent of the cases they identified had been incorrectly treated with anti-tuberculosis medications preceding the diagnosis [1].
Described by Jain as adenomatoid proliferations of the bronchioles that form cysts at the expense of alveoli they are thought to represent an arrest in normal lung growth although the exact aetiology is poorly understood. CCAM were initially classified by Stocker into 3 types. The classification has expanded to include 5 types over more recent years. Type zero CCAM's make up 1-3% of cases, they carry an extremely poor prognosis and a largely incompatible with life. Type I lesions are the most common making up 60-70% of cases. Grossly they are large lesions measuring up to 10 cm in size and are cysts lined by pseudo stratified ciliated cells often interspersed with rows of mucous cells [4]. Type II lesions are smaller and multiple, measuring less than 2 cm. The cysts resemble micro dilated bronchioles separated by normal alveoli and striated muscle. Grossly they are sponge like and solid. Type III CCAM carries a poor prognosis and makes up 5% of cases. They are solid lesions of excessbronchiolar tissue separated by cuboid lined small air spaces. The remaining 15% of cases are type IV and are large cysts lined by flattened epithelium surrounded by loose mesenchymal tissue [4,5]. 60% of type II and type III lesions are associated with other congenital anomalies [6].
The question of what to do with asymptomatic lesions and if acting when to do so is still controversial. Consensus does seem to be moving more decidedly towards surgical management with excision because of the risk of progression to cancer. There is evidence that early surgery, i.e. before the age of 5 years, is beneficial because of increased compensatory capacity and growth of the remaining healthy lung tissue [7]. Type I CCAM is associated with a progression to cancerous lesions [4,8,9]. The incidence of cancer in these lesions is thought to be 1% [4].
Of concern is that these lesions often cannot clinically or radiologically be differentiated from pleuro pulmonary blastoma in its early cystic stages [4]. Another factor in favour of surgical removal is dilemma of exposing children to radiation if the lesions are left in situ given that we know x-ray shows the lesions poorly and that computerised tomography is the modality of choice [4].
Surgery is of course not without its own risks. These include phrenic and recurrent laryngeal nerve injury, chyle leak, haemothorax, bronchopleural fistulas and persistent air leaks, pulmonary emboli, infection, and bleeding [7].There have been studies following conservatively managed children who have been asymptomatic 5 years after birth however the longer term sequelae of these cases is unknown as yet [10]. Our patient experienced no adverse effects from his surgery.
Conclusion
Congenital cystic adenomatoid malformations of the lung are rare and are usually diagnosed in infancy. Our thirty-year-old patient had his lesion excised because of the uncertainty regarding diagnosis and his adverse symptoms however even in asymptomatic patients surgical excision is justified because of the risk of infection and progression to cancer.
References
-
Raman VS, Agarwala S, Bhatnagar V, Panda SS, Gupta AK (2015) Congenital cystic lesions of the lungs: The perils of misdiagnosis - A single-center experience. Lung India 32: 116-118.
-
Jain A, Anand K, Singla S, Kumar A (2013) Congenital cystic lung diseases. J Clin Imaging Sci 3: 5.
-
Juarez-Garcia L, Lopez Rioja Mde J, Leis-Marquez MT, Machuca-Vaca A, Erdmenger-Orellana J (2015) Congenital cystic adenomatoid malformation of the lung, intrauterine diagnostic and treatment. A case report and literature review. Ginecol Obstet Mex 83: 320-327.
-
Dusmet M (2015) Adult lung tumours of childhood origin. Semin Pediatr Surg 24: 196-200.
-
Stocker J (2009) Cystic lung disease in infants and children. Fetal Pediatr Pathol 28: 155-184.
-
Kwak HJ, Moon JY, Kim SI, Kim TH, Sohn JW, et al. (2012) Congenital cystic adenomatoid malformation with bronchial atresia in elderly patients. Tuberc Respir Dis (Seoul) 72: 501-506.
-
Makhija Z, Moir CR, Allen MS, Cassivi SD, Deschamps C, et al. (2011) Surgical management of congenital cystic lung malformations in older patients. Ann Thorac Surg 91: 1568-1573.
-
Harini N, Chakravarthy R, Archana L (2012) Congenital pulmonary airway malformation with mucoepidermoid carcinoma: a case report and review of literature. Indian J Pathol Microbiol 55: 540-542.
-
Hasegawa M, Sakai F, Arimura K, Katsura H, Koh E, et al. (2014) EGFR mutation of adenocarcinoma in congenital cystic adenomatoid malformation/congenital pulmonary airway malformation: a case report. Jpn J Clin Oncol 44: 278-281.
-
Ng C, Stanwell J, Burge DM, Stanton MP (2014) Conservative management of antenatally diagnosed cystic lung malformations. Arch Dis Child 99: 432-437.
