

International Journal of Stem cell Research and Therapy
Murine CD133+CD49flow/+ Cells Derived from ESCs Differentiate into Insulin Producing Cells in vivo
Jesus Ciriza1,3*, Christa Caneda1, Bryce McLelland1 and Jennifer O. Manilay1,2
1Molecular and Cell Biology Unit, University of California, Merced, School of Natural Sciences, USA
2Quantitative and Systems Biology Graduate Group, School of Natural Sciences, University of California, USA
3Biomedical Research Networking Center in Bioengineering, Biomaterials and Nanomedicine, CIBER-BBN, Spain
*Corresponding author: Jesus Ciriza, NanoBioCel Group, Laboratory of Pharmacy and Pharmaceutical Technology, Faculty of Pharmacy, University of the Basque Country UPV/EHU, Vitoria-Gasteiz, 01006, Spain, Tel: +34945014518, E-mail: jeciases@yahoo.es
Int J Stem Cell Res Ther, IJSCRT-1-004, (Volume 1 Issue 1), Research Article; ISSN: 2469-570X
Received: September 18, 2014 | Accepted: December 28, 2014 | Published: December 31, 2014
Citation: Ciriza J, Caneda C, McLelland B, Manilay JO (2015) Murine CD133+CD49flow/+ Cells Derived from ESCs Differentiate into Insulin Producing Cells in vivo. Int J Stem Cell Res Ther 1:004. 10.23937/2469-570X/1410004
Copyright: © 2015 Ciriza J, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Pancreatic progenitors have been identified and isolated during embryonic development showing their potential to differentiate into insulin producing cells after transplantation. Subsequently, they have been proposed as an alternative to the treatment of Type I diabetes. However, fetal pancreata represent a limited donor supply and we proposed that ESC-derivedpancreatic progenitors are another possible option. We isolated committed pancreatic CD133+CD49flow/+ progenitors, expressing neurogenin 3, from insulin producing cells derived from mouse embryonic stem cells in vitro. Here, we demonstrate their potential to differentiate into insulin producing cells in vivo while not forming teratomas. Our results can pave the way for future studies to ascertain the ability of in vitro ESC-derived pancreatic progenitors to be translated for clinical application for Type I diabetes treatments.
Keywords
Murine embryonic stem cells; Pancreatic progenitors; Type I Diabetes; Teratoma; Kidney capsule transplantation; Insulin producing cells
Introduction
Worldwide, 347 million people suffer from diabetes [1] and the World Health Organization projects that diabetes will be the 7th leading cause of death by 2030. In the United States, 25.8 million children and adults had diabetes in 2011, representing 8.3% of the total population [2], and costing $116B for treatment and diagnosis. Type 1 diabetes (TID) is an autoimmune disease that aggressively attacks the insulin producing cells (IPCs) in the pancreas, reducing the number of these cells to levels that force the patient to be dependent on daily insulin injections. Islet cell transplantation can be used for treatment, but sources of autologous and allogeneic donor insulin producing cells for transplantation are limited.
Embryonic stem cells (ESCs) have been proposed as an alternative source of insulin producing cells [3]. Major concerns about the use of ESC-derived cells for therapy include their teratoma formation ability and their possible immune rejection after transplantation. Teratoma formation prevented successful treatment of diabetic mice with ESC-derived cells [4] and immune rejection of insulin producing cells derived from ESCs was observed as early as 3 days post-transplantation into immunocompetent mice [5,6]. To improve this outcome, the enrichment of definitive endodermal progenitors by flow cytometric sorting [7,8] could be used to deplete cultures of undifferentiated cells (which are the main sources of teratomas), while the prediction of the immunogenicity of insulin producing cells derived from ESCs (ESC-IPCs) could indicate possible preconditioning regimens for patients that received ESC-IPC transplants.
Several cell culture protocols have been described for the differentiation of murine nontransgenic [3], but three protocols appear to be the most highly utilized [9-11]. The three protocols share the common initial step of embryoid body (EB) formation, but differ in the use of selection of nestin-expressing cells. When compared side by side, ESC-IPC differentiated using the Blyszczuk et al. [11] protocol were able to produce and release more insulin in vitro than those differentiated by the other two methods. However, they only rescued 33% of diabetic mice within 2 weeks and ultimately, these mice developed teratomas [12], likely due to the remaining undifferentiated ESCs in the cultures at the time of transplantation. Exhaustive microarray studies studying gene expression profiles of ESC-IPC at different stages of in vitro differentiation following the Blyszczuk et al. [11] protocol have shown that murine ESC-derived pancreatic cells appear to be blocked at the embryonic/fetal stage of development [13].
In contrast to fully mature IPCs, pancreatic progenitors generated from ESCs (ESC-PPs) might be more suitable for cellular replacement therapy in TID. A homogenous population of ESC-PPs identified by cell surface markers may not self-renew as stem cells do, but could differentiate into endocrine cells after transplantation [14]. For example, Pdx1+Cxcr4+ ESC-derived cells spontaneously differentiated into IPCs in vivo, and led to a sustained correction of hyperglycemia in diabetic mice for a month with survival rates longer than three months [15]. Late definitive endoderm cells can be identified by the combination of E-cadherin and Decay Accelerating Factor (DAF1/CD55) cell surface markers on cells that co-express also Pdx1 [16] (Figure 1). Ngn3+ cells have been suggested to be another possible murine pancreatic progenitor population [17,18]. CD133+CD49flow/+ cells isolated from murine fetal pancreas have been described to express Ngn3 (Figure 1) as well as insulin-1, glucagon, somatostatin and pancreatic polypeptide after 2 days of co-culture with mitomycin C-treated mouse embryonic fibroblast (MEFs) [8]. Moreover, transplanted CD133+ PDFGR β-cells isolated during murine pancreas development induced the formation of IPCs that expressed Cpeptide (a definitive feature of functional insulin production) within 7 days of culture and showed self-renewal properties after serial transplantation, suggesting they might be true pancreatic stem cells [19]. Ngn3+ cells have been also detected in EBs from murine Ngn3-GFP ESC lines expressing insulin, glucagon and Pdx1, among other pancreatic markers, after sorting and differentiation in vitro [20]. However, the identification with cell surface markers of non-transgenic pancreatic progenitor cells expressing Ngn3 from ESC-differentiation cultures and their application in vivo has not yet been reported.
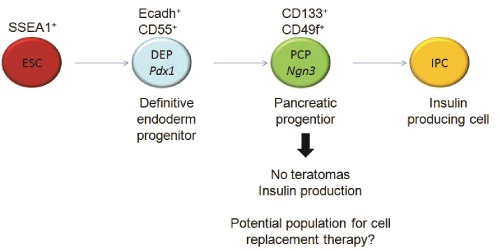
Figure 1: Schematic representation of IPC differentiation: During differentiation from ESC to IPC, definitive endoderm and pancreatic
progenitors can be isolated by means of different extracellular markers
View Figure 1
Here, we present the identification of CD133+CD49flow/+ and E-cadherin+ CD55+pancreatic progenitor cells from a slightly modified ESC-IPC differentiation protocol. We show that CD133+CD49flow/+ ESC-PPs can be transplanted into the kidney capsule of immunodeficient mice, produce insulin, and do not form teratomas.
Results
Heterogeneous and immature ESC-IPCs are generated using current protocols
We initiated our studies with the ESC-IPC differentiation protocol described by Blyszczuk et al. [11] since it seemed to be the most efficient in terms of insulin production and release [12]. This protocol includes the formation and growth of EBs for 5 days, their expansion for 9 days on gelatin-coated plates and their specific differentiation into IPCs for 18 days on collagen I coated plates with a defined differentiationmedium (Figure 2A). Healthy groups of cells could be observed in vitro after Day 5+16, with aslow as 5% FCS in the media (Figure 2A). Gene expression analysis showed similarities between ESC-IPCs and murine pancreatic β islet samples such as expression of insulin 2 and IAPP with very low expression of somatostatin and amylase (Figure 2B). Genes described to be expressedduring the development of insulin producing cells (Glut2, Nestin, Sox17, Isl1 and cytokeratin19 (CK19)) were also detected at different stages of the differentiation protocol including the final stage at Day 5+28 (Figure 2B). However, in contrast to β islet samples, insulin 1was notdetected at any time of differentiation (Figure 2B).
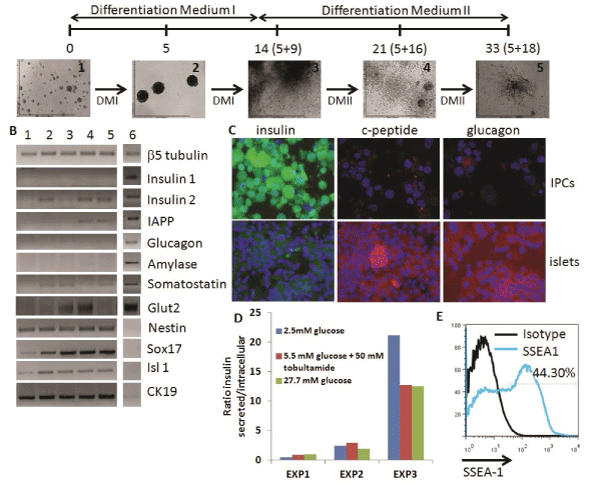
Figure 2: Analysis of ESC-IPCs. A) Scheme of ESC-IPC differentiation protocol. Stages of IPC differentiation: 1: D3-ESC on gelatin,
2: EBs, 3: Day 5+9, 4: Day 5+16, 5: Day 5+28 DMI: Differentiation Medium I and DMII: Differentiation Medium II. B) RT-PCR analysis
of stages of IPC differentiation: 1: D3-ESCs, 2: 5+9, 3: 5+16, 4: 5+28, 5: adult whole pancreas, 6: isolated pancreatic islet. C)
Immunostaining of ESC-IPCs (top row) and islets (bottom row): insulin (green), c-peptide (red), glucagon (red) and nuclei (blue) (20x
magnification). D) Heterogeneous insulin release by IPCs after glucose challenge. E) IPC-ESCs express SSEA-1 (specific marker
staining in blue, isotype control in black)
View Figure 2
Immunohistochemistry analysis showed a high concentration of insulin accumulated inside the IPCs with low expression of c-peptide, in contrast to the β islet samples (Figure 2C). These data suggested that incomplete insulin processing at the protein level was occurring, and indicated that IPCs were not fully functional. Similarly, glucagon expression was much lower in IPCs than β islets (Figure 2C). The insulin release response in IPCs to glucose stimulation invitro was low and heterogeneous, with higher ratios of release at lower glucose concentrations in some experiments (Figure 2D). Finally, we detected that almost half of the IPCs at Day 5+28 of differentiation expressed SSEA-1 (Figure 2E), which suggested that they were teratoma-forming cells [12]. Based on these results, we next considered a different population of cells for further investigation.
Pancreatic progenitors can be detected in ESC-IPC cultures
We investigated if putative pancreatic progenitors could be identified in vitro during differentiation of ESCs into IPCs (Figure 1). First, using flow cytometry, we analyzed the presence of definitive endoderm progenitors with the cell surface marker phenotype. Ecadh+CD55+, as previously described in vitro with other ES cell lines [16]. Low percentages of Ecadh+CD55+ cells were detected at Day 5+16 of differentiation (mean ± SD: 0.93 ± 0.18 %, n=3) (Figure 3A). Almost all Ecadh+CD55+ cells were SSEA-1+, suggesting a high teratoma formation ability potential if transplanted (Figure 3B).
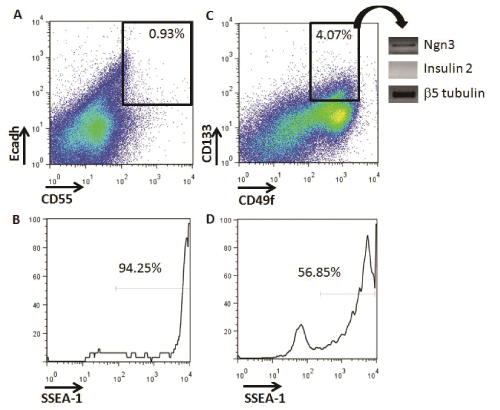
Figure 3: Identification of definitive endoderm and committed pancreatic progenitors from in vitro IPC-ESC cultures by flow cytometry. A) Live cells at
Day 5+16 were analyzed for the expression of E-cadherin and CD55. The gate shows the Ecadherin+CD55+ definitive endoderm progenitors. The data
shown are representative of three independent experiments. B) Histogram analysis of SSEA1 expression on Ecadherin+CD55+ definitive endoderm cells.
C) Live cells at Day 5+16 were analyzed for the expression of CD133 and CD55. The gate shows the CD133+CD49flow/+ committed pancreatic progenitors.
Data shown are representative of three independent experiments. D) Histogram analysis of SSEA1 expression on CD133+CD49flow/+ committed pancreatic
progenitors
View Figure 3
We then searched for pancreatic progenitors that were more differentiated than definitive endoderm progenitors and with less potential of forming teratomas (Figure 1). In vivo, murine and human committed pancreatic progenitors have been identified by the CD133+CD49flow/+ cell surface phenotype, and their potential to differentiate in vitro into insulin producing cells has been verified [8,9]. We were able to quantify an average of 4.07 ± 1.29 % of CD133+CD49flow/+ pancreatic progenitors at Day 5+16 in our IPC differentiation cultures (Figure 3C). This higher frequency of cells, compared to Ecadh+CD55+cells, allowed for the collection of enough cells by flow cytometry to analyze their gene expression. CD133+CD49flow/+ pancreatic progenitors did not express insulin-2, but expressed Ngn3, indicating that they belong to an earlier developmental stage, but were committed to differentiate into IPCs (Figure 3C). These progenitors did not express either pancreatic marker such as Pdx1or Pax4 by RT-PCR (data not shown). CD133+CD49flow/+ pancreatic progenitors also expressed SSEA-1 (Figure 3D). The expression of SSEA-1 indicated their potential to form teratomas, which we then tested in vivo.
CD133+CD49flow/+ pancreatic progenitors did not form teratomas and expressed insulin
First, we transplanted 106 SSEA1+ D3-ESCs under the kidney capsule of NSG mice and all mice developed teratomas by 28 days post-transplantation, as expected (Figure 4A, panel 1). Conversely, as a negative control for teratoma formation, we transplanted 300 islets isolated from adult pancreas and they did not form teratomas (Figure 4A, panel 5). We then tested the ability of our ESC-IPCs to form teratomas after transplantation. First, we confirmed that IPCs at Day 5+28 of differentiation expressed insulin 2, and then we transplanted 4-6x106 IPCs. All of the IPC-recipient mice developed teratomas by 28 days after transplantation (Figure 4A, panel 2). This result confirmed previous results [11] and justified the exploration of an alternative ESC-derived cell population for the replacement of insulin.
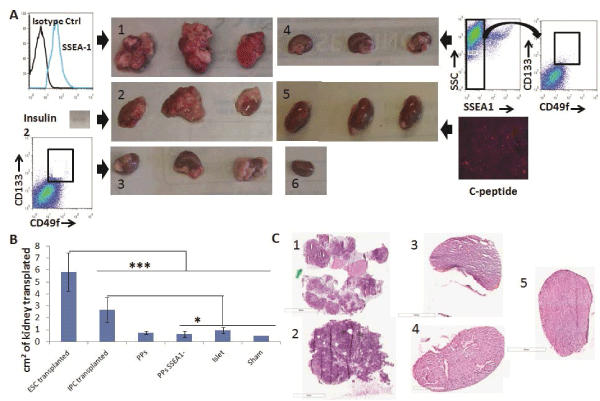
Figure 4: Engraftment and teratoma formation analysis of ESC-IPCs and ESC-PPs in vivo. A) Macroscopic observations of kidneys on Day 28 after
transplantation of: 1: 106 ESCs; 2: 6x106 IPC-ESCs (expressing insulin 2, inset); 3: 5x105 CD133+CD49flow/+ committed pancreatic progenitors; 4: :5x105
CD133+CD49flow/+SSEA1- committed pancreatic progenitors; 5: 300 islets (positive for c-peptide); and 6: no cells (sham control). B) Comparison of kidney
sizes after transplantation (*: p< 0.05 and ***: p< 0.001). C) Hematoxylin/eosin staining from kidney sections transplanted with: 1: 106 ESCs; 2: 6x106 IPC-ESCs;
3:5x104 CD133+CD49flow/+ committed pancreatic progenitors; 4: 5x104 CD133+CD49flow/+SSEA1- committed pancreatic progenitors and 5: 300 islets
View Figure 4
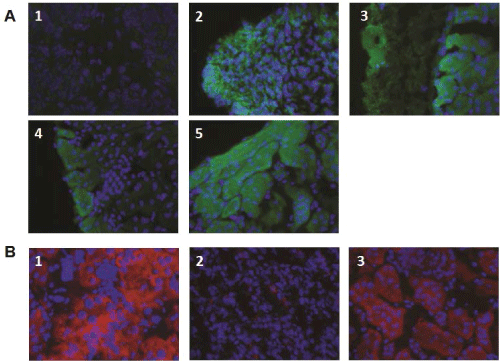
Figure 5: Insulin and c-peptide expression in kidneys transplanted with ESC-IPCs and ESC-PPs. (A) Kidney sections were stained for insulin from mice
transplanted with: 1: 106 ESCs; 2: 6x106 IPC-ESCs; 3:5x105 CD133+CD49flow/+ committed pancreatic progenitors; 4: 5x104 CD133+CD49flow/+SSEA1- committed
pancreatic progenitors; and 5: 300 islets. (B) Kidney sections were stained for c-peptide from mice transplanted with: 1: 5x104 CD133+CD49flow/+ committed
pancreatic progenitors; 2: 5x104 CD133+CD49flow/+SSEA1-
View Figure 5
We hypothesized that more immature pancreatic progenitors could be generated, isolated and transplanted in vivo, and that after transplantation, they would not form teratomas and would produce insulin. As mentioned above, CD133+CD49flow/+ cells have been demonstrated to be pancreatic progenitors (PPs) in mouse embryos [8] and we confirmed that CD133+CD49flow/+ cells were generated in our in vitro cultures. We tested the function and potential of these CD133+CD49flow/+ ESC-PPs to form teratomas in vivo. Since SSEA-1 expression is often equated with pluripotency and teratoma-forming ability in ESCs, we also compared the behavior of sorted SSEA-1- CD133+CD49flow/+ ESC-PPs. We transplanted 5x104pancreatic progenitors (SSEA1+ depleted or not) as previously described [8]No teratomas were observed in mice transplanted with either population (Figure 4A, panels 3,4). No significant differences were found amongst the sizes of the kidneys transplanted with ESC-PPs, islets or sham controls (Figure 4B), a result that correlated with a lack of teratoma forming ability in ESC-derived PPs. The absence of teratoma formation in recipients of CD133+CD49flow/+ or SSEA-1- CD133+CD49flow/+ ESC-PP cells was confirmed by histological analysis of kidney sections from the transplanted mice (Figure 4C, panels 3,4). In contrast, teratomas were clearly observed in kidneys transplanted with ESCs and ESC-IPCs (Figure 4C, panels 1,2). ESC-IPCs, CD133+CD49flow/+ ESC-PPs, SSEA-1- CD133+CD49flow/+ ESC-PPs and islets exhibited positive staining for insulin (Figure 5A, panels 2-5), whereas sections of kidney capsules transplanted with ESCs did not (Figure 5A, panel 1). Insulin production was confirmed via the observation of strong c-peptide expression in the kidneys thatwere transplanted with CD133+CD49flow/+ ESC-PPs and control islets (Figure 5B, panels 1,3). SSEA-1-CD133+CD49flow/+ ESC-PPs showed positive, but lower expression of c-peptide (Figure 5B, panel 2).
Discussion
We generated ESC-IPCs that express endocrine genes such as insulin 2, glucagon and somatostatin, and our data showed that ESC-IPC cultures heterogeneously secrete insulin protein, corroborating the findings of other groups [12]. Based on the lack of peptide signal by immunohistochemistry, we posit that the insulin is not completely processed intracellularly within ESC-IPCs and consequently, is not released properly outside the cell. One possible reason for this non-functional insulin release is incomplete differentiation of the ESCIPCs, resulting in a cell that is devoid of fully developed mechanisms for insulin processing. Other groups have suggested that the detection of insulin from ESC-IPCs is an artifact from apoptotic or necrotic cells [21] or a mixture of de novo insulin production mixed with insulin uptake from the medium [22] and this controversy is still unresolved. In our hands, insulin release from ESC-IPCs after glucose challenge varied widely from experiment to experiment, perhaps reflecting the presence of a heterogenous mixed of cells that are generated in each independent ESC-IPC culture [12]. The immature nature of ESC-IPCs, their inconsistent release of insulin, and their propensity to form teratomas precludes their clinical translation for the treatment of hyperglycemia in diabetic patients.
In contrast, we confirmed that ESC-derived CD133+CD49flow/+ pancreatic progenitor cells did not form teratomas after 28 days of in vivo transplantation. This was surprising, as 50% of CD133+CD49flow/+ cells expressed SSEA1, which is often associated with pluripotency. It has been previously hypothesized that human pancreas cells that are positive for stage-specific embryonic antigen 4 (SSEA4) co-express ductal pancreatic progenitor markers and can be differentiated in vitro to pancreatic hormone-expressing cells [23]. We hypothesize that SSEA1 could be a murine marker for pancreatic progenitors, sharing similarities to human SSEA4 as a murine ductal pancreatic progenitor marker. However, after transplantation in vivo, SSEA-1-CD133+CD49flow/+ ESC-PPs expressed lower c-peptide levels than CD133+CD49flow/+ ESC-PPs, a result which suggests that some pancreatic progenitors may be depleted if the SSEA1 marker is used for isolation of PPs. Regardless of the expression of SSEA1, both unfractionated CD133+CD49flow/+ and sorted CD133+CD49flow/+SSEA1+populations did not form teratomas in vivo.
It is possible that the lack of teratoma-formation we observed is simply due to the transplantation of insufficient numbers of cells required to form teratomas in vivo. We do not feel this is the case, as we selected our cell dosage based on previous studies in which the same minimal cell dose resulted in 100% teratoma-forming ability after transplantation into immunodeficient mice [8,24,25]. The lack of teratoma-forming ability and the clear expression ofinsulin and C-peptide in both unfractionated CD133+CD49flow/+ and sortedCD133+CD49flow/+SSEA1+ populations lead us to conclude that PPs are a potentially feasible cellular therapy.
One challenge to the use of ESC-derived cells is that in vitro differentiation of ESCs to specific cell lineages may not necessarily produce cells that express the same cell surface markers as their in vivo-derived counterparts [26]. To our knowledge, our study is the first to identify murine pancreatic progenitor populations derivedfrom ESCs in culture, and we have confirmed that ESC-derived CD133+CD49flow/+ cells arefunctionally similar to their in vivo counterparts in mouse embryos. Clearly, improvements in the generation of higher yields of pancreatic progenitor cells are required for practical application, and this could be achieved by discovery of methods to block unwanted lineage differentiation, as well as scaled up production and isolation by means of bioreactors and other stem cell separation technologies [27].
In conclusion, we have identified mouse ESC-derived pancreatic progenitor cells in vitro and discovered that they can survive and produce insulin in vivo. We envision that our phenotypic and functional data could be further utilized to study and improve the long-term survival of in vitro derived pancreatic progenitor cells and their ability to produce insulin in sufficient quantities to alleviate hyperglycemia in humans.
Materials and Methods
Mice
NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ(NSG) and 129S2/SvPasCrl (129) mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA) and Charles River (Wilmington, MA, USA) respectively, or bred in house and housed in sterile microisolator cages with sterile feed and autoclaved water. Mice were euthanized by CO2asphyxiation or by cervical dislocation in the case of pancreatic islet isolation. All procedures were approved by the UC Merced Institutional Animal Care and Use Committee (IACUC).
Antibodies
The following monoclonal antibodies were purchased from BioLegend (San Diego, CA) and used in flow cytometry: PE anti-mouse CD55 (DAF) (RIKO-3), PE Armenian Hamster IgG Isotype Ctrl (HTK888), APC anti-human/mouse CD49f Antibody (GoH3), APC Rat IgG2a, ?Isotype Ctrl Antibody (RTK2758), biotin anti-mouse/human SSEA-1 (MC-480) and biotin mouse IgM, κ Isotype Ctrl (MM-30). Anti-Mouse CD133 (Prominin-1) PE (13A4), Rat IgG1 KIsotype Control PE (eBRG1), anti-CD324 (E-Cadherin) Alexa Fluor® 647 (DECMA-1) and Rat IgG1 K Isotype Control Alexa Fluor® 647 (eBRG1) were purchased from eBioscience (San Diego, CA). Fc receptors were blocked with anti-CD16/32 (2.4G2) purchased from eBioscience(San Diego, CA). Biotin labeled antibodies were stained with streptavidin (SA)-PE, SA-PE-Cy7 or SA-PE-Cy5 purchased from Biolegend (San Diego, CA). The following polyclonal antibodies were used for immunohistochemistry: guinea pig polyclonal to insulin (Abcam, Cambridge, MA, USA), anti-c-peptide and anti-glucagon (Cell Signaling Technology, Danvers, MA, USA), donkey anti guinea pig IgG Fluorescein conjugated (Millipore, Billerica, MA, USA) and goat anti-rabbit IgG-Atto 633 (Sigma-Aldrich, St. Louis, MO). Samples were previously blocked with normal goat serum purchased from Jackson Immunoresearch Laboratories, Inc. (West Grove, PA, USA).
Embryonic stem cell differentiation and culture
The D3 embryonic stem cell line (ES-D3) from ATCC (Manassas, VA, USA) was cultured in undifferentiated state by subculture every 2-3 days on confluent feeder layers (STO cells) treated with 10 μg/ml of mitomycin C (Fisher, Pittsburg, PA, USA) in a medium containing 103 units/ml LIF (Millipore, Billerica, MA, USA), 0.15 mM monothioglycerol (MTG), 100 units/ml penicillin 100 μg/ml streptomycin (Invitrogen, Grand Island, NY, USA), 15% FCS (lot # A50A01X, Gemini Bio Products, West Sacramento, CA, USA) and DMEM (Invitrogen, Grand Island, NY, USA). STO cells were removed by plating ES-D3 and STO cells cocultures on gelatin coated plates for 2 days in medium containing 103 units/ml LIF, 0.15mM MTG, 100 units/ml penicillin 100 μg/ml streptomycin, 15% FCS and Iscove's modified Dulbecco's medium (IMDM, Invitrogen, . Grand Island, NY, USA).
Cells were differentiated in embryoid bodies (EBs, 600 ESCs/EB) by hanging drops for 2 days, further growing EBs for 3 days on suspension in IMDM supplemented with supplemented with 20% FCS (lot # A0039, Atlanta Biologicals, Lawrenceville, GA, USA), 2mM L-glutamine (Invitrogen, Grand Island, NY, USA), 0.1 mM essential amino acids (Invitrogen, Grand Island, NY, USA), 0.15 mM MTG, 100 units/ml penicillin and 100 μg/ml streptomycin. Next, EBs were cultured on gelatin coated plates with the same medium for 9 days changing it every 2-3 days. At day 5+9 of gelatin-attached EBs and their outgrowths were dissociated with 0.25 % trypsin-EDTA (Invitrogen) and re-plated onto collagen I-coated tissue culture dishes. Differentiation medium consisted of DMEM/F12 (Invitrogen) supplemented by 20nMprogesterone, 80 μM putrescine, 1 μg/ml laminin, 10 mM nicotinamide, 25 μg/ml insulin, 30nM sodium selenite, 50 μg/ml transferrin, B27 media supplement (dilution 1:50) (Invitrogen), 100 units/ml penicillin, 100 μg/ml streptomycin and 10% FCS (lot # A0039, Atlanta Biologicals, Lawrenceville, GA, USA). All the reagents were ordered from Sigma-Aldrich (St. Louis, MO) unless specified differently. At Day 5+10, medium was changed and cells were cultivated in the same differentiation medium but 5% FCS, changing medium every 2 days until they were used. Cultures were stopped at Day 5+28.
Gene expression analysis
RNA from ESCs, EBs at Day 5+9 and differentiated cells at Days 5+16 and 5+28 was extracted using RNeasy® Protect Mini Kits (Qiagen, Valencia, CA) and from sorted pancreatic progenitors using Trizol (Invitrogen, NY), following the manufacturer's instructions. RNA samples were treated with DNAseI (Roche, San Francisco, CA) and the concentration of RNA was determined using a Nanodrop spectrophotometer (ThermoScientific, Waltham, MA). Next, cDNA was synthesized with SuperScriptIII First-Strand Synthesis System for RT-PCR Kit (Invitrogen, Grand Island, NY, USA). The gene expression of 17 genes was analyzed by PCR as previously described with β5-tubulin as the housekeeping gene [28]. For Ngn3, Pdx1 and Pax4 gene expression analysis, clones containing the respective gene were purchased from Thermo Scientific (Waltham, MA, USA) and used as positive controls. PCR products were visualized after electrophoresis in ethidium bromide stained 2% agarose gels with a BioRadChemiDoc imaging system (BioRad, Hercules, CA, USA). Sizes of PCR products were compared with a 100 bp Low Scale DNA ladder (Fisher, Pittsburg, PA, USA).
Immunohistochemistry
Immunohistochemistry was performed on IPC-ESC grown on six well-plates at Days 5+16 or 5+28 of differentiation. Cells were collected by trypsinization and cytospun on slides (3x105 cells/slide) with a Shandon Cytospin III cytocentrifuge at 1000 rpm for 8 minutes. INS-1 832/13 insulinoma cell line cultures (a gift from Dr. Chris Newgard, Duke University) were also prepared as controls for insulin expression. Samples were fixed with 4% paraformaldehyde (PFA) for 15 minutes at room temperature, rinsed 3 times with phosphate buffered saline (PBS) and blocked for 30 minutes at room temperature with blocking buffer consisting of 10% normal goat serum and 0.1 % Triton®-X-100 in PBS. After blocking, samples and controls were incubated at 4°C overnight with guinea pig anti-insulin, rabbit anti-c-peptide or rabbit antiglucagon. Samples stained with secondary antibodies alone were prepared as negative controls.The next day; samples were rinsed three times with PBS, incubated for 45 minutes at 37°C with anti-guinea pig IgG FITC or anti-rabbit IgG Atto 633, and stained with 0.5 μg/ml DAPI. Finally, samples were sealed with Vectashield mounting medium (Vector, Burlingame, CA , USA) and visualized by fluorescence microscopy, using the Olympus BX51 Microscope, the 10x/0.30 and 40x/0.75 U Plan FLN lenses and the Image-Pro Plus 5.0 software.
Insulin release in vitro
At Day 5+28 of differentiation, IPC-ESC were cultured with insulin-free differentiation medium for 48 hours prior to challenge cells with different concentrations of glucose. Medium was changed again 3 hours before glucose challenge and cultures were washed five times with PBS. Cells were further incubated for 90 minutes with 2.5 mM glucose in Krebs Ringer bicarbonate HEPES (KRBH) buffer consisting of 118 mM NaCl, 4.7 mM KCl, 1.1 mM KH2PO4, 25 mM NaHCO3, 3.4 mM CaCl2, 2.5 MgSO4, 10 mM HEPES, and 2 mg/ml BSA at 7.4 pH. ESIPCs were challenged with KRBH containing 2.5 mM glucose, 5.5 mM glucose and 50 μM tolbutamide, or 27.7 mM glucose for 1 hour. The supernatants from the different cultures were collected and cells were dissociated by trypsinization and resuspended in acid ethanol for protein extraction. Upon resuspension, cells were sonicated and incubated overnight at 4°C. Next day, acid ethanol suspension was neutralized with 1 M Tris Buffer, pH 7.5. Total protein content was determined by Bradford assay with the Coomassie Protein Assay Kit (ThermoScientific, Waltham, MA). Released and intracellular insulin levels were measured by means of ELISA with the Mercodia Ultrasensitive Mouse Insulin Enzyme-Linked I mMunosorbent Assay (Mercodia Developing Diagnostics, Uppsala, Sweden) according to manufacturer instructions.
Flow cytometry analysis and sorting
Samples containing 106 single cells from ES or ES-IPCs cells at stages 5+16 or 5+28 were blocked with anti-CD16/32 for 5 minutes at 4°C. Next, they were stained for 15 minutes at 4°C with either an antibody cocktail containing anti CD133-PE, anti CD49f -APC and biotin anti SSEA-1, or the cocktail containing anti E-Cadherin-Alexa Fluor® 647, anti CD55- PE and biotin anti SSEA-1. For each antibody cocktail, "fluorescence minus-one" or fluorescently-labeled isotype antibody controls were also prepared. Finally, cells were resuspended in 0.1 μg/ml DAPI before sorting to exclude dead cells. Cells were analyzed or isolated using a FACS Aria flow cytometer (Becton Dickinson, Franklin Lakes, NJ). Single color samples for dye compensation were prepared for each experiment. For each study, three independent experiments were performed.
Pancreatic islet isolation
After cervical dislocation, pancreata from 129 mice were infused with 3 ml/mouse of 0.8 mg/ml collagenase P solution (Roche Applied Science, Indianapolis, IN, USA), removed and digested for at least 16 minutes at 37oC in the same collagenase P solution. Cell suspensions were filtered through a 70 micron cell strainer and washed several times with a solution consisting of 10 mM HEPES (Fisher, Pittsburg, PA, USA), 10 μg/μl Dnase I (Roche Applied Science, Indianapolis, IN, USA), 100 units/ml penicillin, 100 μg/ml streptomycin, 1.7 mM CaCl2 (Fisher, Pittsburg, PA, USA) and 1.2 mM MgCl (Fisher, Pittsburg, PA, USA) in HBSS (Invitrogen), until clumps were not visible in the supernatant. Cells were resuspended in 5 ml of Histopaque-1119 (Sigma-Aldrich, St. Louis, MO) and slowly added 5 ml of culture medium consisting of 25 mM HEPES, 0.1 mM non-essential amino acids (Invitrogen), 100 units/ml penicillin, 100 μg/ml streptomycin, 10% FBS in RPMI 1640 (Invitrogen) on top of the Histopaque solution. Islets in the Histopaque-culture medium interface were collected and 2000 rpm for 20 minutes without brake spinning, washed three times with washing medium, and resupended in 5 ml of culture medium to further being collected by hand picking under a dissecting microscope [29].
In vivo engraftment
Eight to 10 week-old NSG mice were anesthetized with isofluorane and shaved. A vertical incision through dermal layers and peritoneum was made and the left kidney pulled out. After a small incision in the kidney capsule, 300 pancreatic islets, 4-5x106 ESC-IPCs, 5x104pancreatic progenitors (SSEA1+ depleted or not) or 106 ESCs were inserted under the capsule. The kidney capsule was cauterized, the peritoneum sutured and skin stapled after transplantation. The post-operative analgesic buprenorphine was injected at doses of 0.05-0.1 mg/kg immediately after surgery and 18 hours later. Kidneys were collected 28 days post-transplantation and grafts were assessed by visualization and measurement of their diameter as well as histology. Sections between 5 to 6 microns from transplanted and non-transplanted kidneys were prepared at the UCSF Mouse Pathology Core for a fee. Immunohistochemistry was performed as described previously above.
Data analysis
Flow cytometry data was analyzed using FlowJo software, version 7.2.4 (TreeStar) or version 8.8.7 (TreeStar). Fluorescence microscopy analysis was performed with Image-Pro Plus5.0 software, choosing randomly the representative micrographs from all samples in each experiment. Tukey and Student t-test statistical analysis were performed using SPSS software, 20 Version 13.0. Data are expressed as means ± standard deviation. P = 0.05 and p = 0.001 were considered significant for comparison of groups using Tukey test.
Acknowledgments
We thank Mr. Roy Hoglund, Ms. Emily Slocum and the UC Merced Department of Animal Research Services staff for excellent mouse husbandry and animal care. We also thank Mr. Gregory L. Szot from the UCSF Diabetes Center for training in aseptic islet isolation and kidney capsule transplantation techniques. This work was supported by the California Institute for Regenerative Medicine (RN1-00554-1).
References
-
Danaei G, Finucane MM, Lu Y, Singh GM, Cowan MJ, et al. (2011) National, regional, and global trends in fasting plasma glucose and diabetes prevalence since 1980: systematic analysis of health examination surveys and epidemiological studies with 370 country-years and 2.7 million participants. Lancet 378: 31-40.
-
Atlanta, G.U.S.D.o.H.a.H.S., Centers for Disease Control and Prevention. (2011) Centers for Disease Control and Prevention. National diabetes fact sheet: national estimates and general information on diabetes and prediabetes in the United States, 2011.
-
Bonner-Weir S, Weir GC (2005) New sources of pancreatic beta-cells. Nat Biotechnol 23: 857-861.
-
Fujikawa T, Oh SH, Pi L, Hatch HM, Shupe T, et al. (2005) Teratoma formation leads to failure of treatment for type I diabetes using embryonic stem cell-derived insulin-producing cells. Am J Pathol 166: 1781-1791.
-
Boyd AS, Wood KJ (2009) Variation in MHC expression between undifferentiated mouse ES cells and ES cell-derived insulin-producing cell clusters. Transplantation 87: 1300-1304.
-
Boyd AS, Wood KJ (2010) Characteristics of the early immune response following transplantation of mouse ES cell derived insulin-producing cell clusters. PLoS One 5: e10965.
-
Yasunaga M, Tada S, Torikai-Nishikawa S, Nakano Y, Okada M, et al. (2005) Induction and monitoring of definitive and visceral endoderm differentiation of mouse ES cells. Nat Biotechnol 23: 1542-1550.
-
Sugiyama T, Rodriguez RT, McLean GW, Kim SK (2007) Conserved markers of fetal pancreatic epithelium permit prospective isolation of islet progenitor cells by FACS. Proc Natl Acad Sci U S A 104: 175-180.
-
Lumelsky N, Blondel O, Laeng P, Velasco I, Ravin R, et al. (2001) Differentiation of embryonic stem cells to insulin-secreting structures similar to pancreatic islets. Science 292: 1389-1394.
-
Hori Y, Rulifson IC, Tsai BC, Heit JJ, Cahoy JD, et al. (2002) Growth inhibitors promote differentiation of insulin-producing tissue from embryonic stem cells. Proc Natl Acad Sci U S A 99: 16105-16110.
-
Blyszczuk P, Asbrand C, Rozzo A, Kania G, St-Onge L, et al. (2004) Embryonic stem cells differentiate into insulin-producing cells without selection of nestin-expressing cells. Int J Dev Biol 48: 1095-1104.
-
Boyd AS, Wu DC, Higashi Y, Wood KJ (2008) A comparison of protocols used to generate insulin-producing cell clusters from mouse embryonic stem cells. Stem Cells 26: 1128-1137.
-
Rolletschek A, Schroeder IS, Schulz H, Hummel O, Huebner N, et al. (2010) Characterization of mouse embryonic stem cell differentiation into the pancreatic lineage in vitro by transcriptional profiling, quantitative RT-PCR and immunocytochemistry. Int J Dev Biol 54: 41-54.
-
Zhou Q, Brown J, Kanarek A, Rajagopal J, Melton DA (2008) In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature 455: 627-632.
-
Raikwar SP, Zavazava N (2011) Spontaneous in vivo differentiation of embryonic stem cell-derived pancreatic endoderm-like cells corrects hyperglycemia in diabetic mice. Transplantation 91: 11-20.
-
Shiraki N, Harada S, Ogaki S, Kume K, Kume S (2010) Identification of DAF1/CD55, a novel definitive endoderm marker. Cell Struct Funct 35: 73-80.
-
Gu G, Wells JM, Dombkowski D, Preffer F, Aronow B, et al. (2004). Global expression analysis of gene regulatory pathways during endocrine pancreatic development. Development 131: 165-179.
-
Mellitzer G, Martin M, Sidhoum-Jenny M, Orvain C, Barths J, et al. (2004) Pancreatic islet progenitor cells in neurogenin 3-yellow fluorescent protein knock-add-on mice. Mol Endocrinol 18: 2765-2776.
-
Hori Y, Fukumoto M, Kuroda Y (2008) Enrichment of putative pancreatic progenitor cells from mice by sorting for prominin1 (CD133) and platelet-derived growth factor receptor beta. Stem Cells 26: 2912-2920.
-
Ku HT, Chai J, Kim YJ, White P, Purohit-Ghelani S, et al. (2007) Insulin-expressing colonies developed from murine embryonic stem cell-derived progenitors. Diabetes 56: 921-929.
-
Hansson M, Tonning A, Frandsen U, Petri A, Rajagopal J, et al. (2004) Artifactual insulin release from differentiated embryonic stem cells. Diabetes 53: 2603-2609.
-
Paek HJ, Morgan JR, Lysaght MJ (2005) Sequestration and synthesis: the source of insulin in cell clusters differentiated from murine embryonic stem cells. Stem Cells 23: 862-867.
-
Afrikanova I, Kayali A, Lopez A, Hayek A (2012) Is stage-specific embryonic antigen 4 a marker for human ductal stem/progenitor cells? Biores Open Access 1: 184-191.
-
Lee AS, Tang C, Cao F, Xie X, van der Bogt K, et al. (2009) Effects of cell number on teratoma formation by human embryonic stem cells. Cell Cycle 8: 2608-2612.
-
Prokhorova TA, Harkness LM, Frandsen U, Ditzel N, Schrøder HD, et al. (2009) Teratoma formation by human embryonic stem cells is site dependent and enhanced by the presence of Matrigel. Stem Cells Dev 18: 47-54.
-
McKinney-Freeman SL, Naveiras O, Yates F, Loewer S, Philitas M, et al. (2009) Surface antigen phenotypes of hematopoietic stem cells from embryos and murine embryonic stem cells. Blood 114: 268-278.
-
Zhu B, Murthy SK (2013) Stem Cell Separation Technologies. Curr Opin Chem Eng 2: 3-7.
-
Schroeder IS, Rolletschek A, Blyszczuk P, Kania G, Wobus AM (2006) Differentiation of mouse embryonic stem cells to insulin-producing cells. Nat Protoc 1: 495-507.
-
Szot GL, Koudria P, Bluestone JA (2007) Murine pancreatic islet isolation. J Vis Exp: 255.
