Myelodysplastic syndromes (MDS) are myeloid neoplasms characterized by peripheral blood cytopenias, with associated morphologic dysplasias, and recurrent cytogenetic abnormalities. However, approximately 40-50% of MDS have no detectable cytogenetic abnormalities or cytogenetically-normal (CN-MDS). MDS cases with concurrent cytogenetics/FISH and molecular testing were identified from a two-year cohort with a median time to follow-up of 506 days. A total of 153 MDS were gathered, and we identified 58 patients with CN-MDS. Somatic mutations were seen in 93% of the CN-MDS patients, involving a variety of cellular pathways. Interestingly, acquired mutations involving genes associated with histone modifier and signaling pathways at initial diagnosis significantly show an association with increased risk of progression to acute myeloid leukemia (AML). Multivariate analysis confirmed that mutations involving these pathways conferred significant independent factors with respect to AML progression. Overall survival was also significantly shorter in those CN-MDS with signaling gene mutations.
Myelodysplastic syndromes (MDS) are heterogeneous group of diverse clonal hematopoietic stem cell disorder defined by the World Health Organization (WHO) as the presence of peripheral blood cytopenias, dysplasias in one or more of the major myeloid lineages, ineffective hematopoiesis, recurrent cytogenetic abnormalities and increased risk of developing acute myeloid leukemia (AML) [1]. While cytopenias and morphologic dysplasias are not pathognomonic to MDS, other causes of dyspoiesis are needed to be essentially excluded (including nutritional etiologies, toxin or drug/radiation exposures, vitamin deficiencies, infectious agents, alcohol abuse, autoimmune disorders, chronic kidney and liver diseases, and endocrinopathies) before diagnosing MDS [2].
Results from cytogenetic studies by conventional karyotyping and fluorescence in-situ hybridization (FISH) assays serve as important parameters included in the revised international prognostic scoring system (IPSS-R) score in MDS, which is proven to be beneficial for determination of prognostic status of untreated patients with disease [3]. The lower risk MDS patients are defined as those having a risk of very low, low, or intermediate disease according to the IPSS-R with a score of ≤ 3.5 points [4]. The comprehensive cytogenetic scoring system (CCSS) was adapted by the WHO which identifies specific prognostic stratification of MDS based on the cytogenetics clone. Overall survival was reported to be significantly different independently on the cytogenetics status [5].
It was reported that chromosomal defects are observed in approximately 50% of primary and 80% of therapy-related MDS (t-MDS) cases [6,7]. It was also evident that prognosis of patients deteriorates with increasing number of cytogenetic abnormalities, suggesting the clonal evolution and genetic instability of MDS clone [5]. The WHO has already defined the specific recurrent balanced and unbalanced chromosomal abnormalities, which are considered presumptive evidence of MDS, even in the absence of definite morphological dysplastic features [1].
Most cytogenetics and molecular markers, although being useful in the diagnosis and prognostication of MDS, are not specific for MDS, and are also found in other bone marrow neoplasms [2]. Somatic mutations are not officially integrated in the diagnostic criteria of MDS in the revised WHO classification, yet they serve as supportive evidence, in the proper clinical context, and marrow morphologic findings [8]. Detection of an MDS-associated mutation can provide additional diagnostic support in cases of which the clinical presentation and morphologic features are ambiguous [9].
While large number of cases are cytogenetically abnormal MDS (CA-MDS), significant proportion of cases are recognized to be cytogenetically-normal MDS (CN-MDS). Recurrent cytogenetic abnormalities provide prognostic information in MDS; however 50-70% of MDS are CN-MDS, and some overlapping morphological and/or pathophysiological features make it challenging to differentiate between MDS and other diseases/disorders [10,11]. We hypothesized that patterns of coexisting mutations in CN-MDS can aid in the diagnostic utility and can further aid in risk stratification of CN-MDS, and provide insight to the disease biology and progression to AML, using next-generation sequencing (NGS).
This study describes the mutational landscape of cytogenetic risk-stratified MDS, and investigates the co-mutations in providing information on the prognostic impact in the progression of CN-MDS. We thought that next-generation sequencing (NGS) could provide a comprehensive mutational profile to determine a clonal abnormality in CN-MDS. We herein report NGS mutational analysis findings of CN-MDS and further compare their mutational spectrum from the different risk-stratified subgroups based on the CCSS classification (Table 1).
Table 1: CCSS prognostic stratification of MDS. View Table 1
All MDS cases from the Oregon Health & Science University (OHSU) were obtained from January 2018 to December 2019 through the laboratory information system pathology search tool, upon approval of the OHSU Institutional Review Board. A total of 153 patients were gathered with complete cytogenetic results, molecular studies, and histopathology reports at initial diagnosis. All those with incomplete laboratory MDS work up were excluded, whereas cases of therapy-related MDS (t-MDS) and MDS cases progressing to AML or secondary AML (s-AML) were included in the study. The patient cohort consisted of 89 males and 64 females (M:F ratio 1.4:1) with median age of diagnosis at 67 years. Data from transformed AML cases were also recorded based on follow-up pathology reports. Median time of follow-up is 506 days (Table 2).
Table 2: Patient characteristics. View Table 2
Standard cytogenetics karyotype analysis was performed on peripheral blood or bone marrow aspirate/biopsy material. At least 20 metaphase cells were examined for each case. FISH analysis was also performed using the following probes: 5q EGR1 (5q31), D7S486 (7q31), CEP 8 (SA), D20S108 (20q12), MLL (11q23) break-apart, and TP53 (17p13.1).
NGS was designed to detect alterations in a panel of 220 genes, many of which are known to play a role in leukemia and lymphoma pathogenesis, diagnosis, prognosis, response to therapy, disease monitoring, or inherited predisposition. All or selected coding exons and the canonical splice sites of genes are sequenced.
Briefly, genomic DNA was extracted and purified from blood, bone marrow or other hematopoietic tissue from fresh or fixed samples. If the submitted sample is from formalin fixed-paraffin embedded tissues (FFPE), the specimen is examined microscopically and, if deemed helpful to enhance sensitivity, genomic DNA is extracted and purified from micro dissected, tumor-rich areas. Mutations were screened by massively parallel sequencing NGS using a combination of multiplexed PCR (customized QIAseq targeted DNA panel with molecular barcodes) and sequencing on an Illumina platform (NextSeq 500 or 550). Sequencing data is then aligned against a reference genome [hg19]. An in-house bioinformatics analysis pipeline was used that employs multiple established variant calling tools (FreeBayes, MuTect2, and Scalpel) and variant annotation tools (Oncotator). The assay is validated according to AMP guidelines [12,13].
With regard to insertions and deletions, NGS is known to be biased toward detecting shorter alterations, and often to underestimate variant allele frequency (VAF) of these types of variants. Therefore, a supplementary non-biased size-based assay is concomitantly run to ensure that internal tandem duplication insertions in FLT3 exon 14 are not missed. Furthermore, a supplementary assay is concomitantly run to detect partial tandem duplications in KMT2A/MLL (aka MLL-PTD) if warranted by the clinicopathological findings of the patient.
A laboratory-developed algorithm was used for variant annotation against public databases such as COSMIC (http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/) and dbSNP (http://www.ncbi.nlm.nih.gov/SNP/). Genomic variants have been classified in accordance with the 2017 standards and guidelines recommended by AMP/ASCO/CAP and currently available resources [14].
The low limit of detection for this assay is 2% VAF at a minimal 900 read depth (5% at 700 read depth and 7% at 500 read depth). However, a small fraction (100 minus the percentage in the parenthesis) of the targeted regions of the following genes: ASXL1 (99%), MED12 (99%), PCLO (99%), DNMT3A (99%), SPEN (99%), NOTCH2 (99%), JAK1 (99%), ARID1B (99%), SETBP1 (98%), WAS (98%), DTX1 (98%), RAD21 (98%), CD79A (97%), STAT5B (94%), SPI1 (94%), BRD4 (93%), PMS2 (89%) have a higher low limit of detection of 10-15% VAF (if less than 250 read depth), or could harbor mutations that were missed by this analysis if the read depth is below 100.
The Fisher exact test was used to compare differences in the mutational profile of genes identified in MDS cases. All p-values were calculated with the use of two-sided tests. A p-value of < 0.05 was considered statistically significant. AML transformation and overall survival (OS) were calculated from the date of MDS diagnosis to the date of AML diagnosis and date of death or last patient contact respectively. Data were censored at the time of patients last known to be alive. Patient survival was analyzed using the Kaplan-Meier method and compared using log rank test. Common/frequent mutations were defined as presence of 2% or more mutations per case. Multivariate logistic regression analysis was performed for AML transformation versus sex, age, treatment protocol, transplant history, and mutational status (Table 3). Statistical analyses were performed using GraphPad Prism (version 8.3.1, GraphPad Software, San Diego, CA, USA) and Stata software (version 14.2, StataCorp LLC, College Station, Texas, USA).
Table 3: Logistic regression multivariate analysis for CN-MDS. CN-MDS patients with mutations in histone modifier genes and signaling genes have 20- and 48-fold higher odds, respectively, of transforming to AML than patients without these mutations; whereas sex, age, treatment regimen, and transplant history are not significant contributing factors. View Table 3
Based on the CCSS prognostic subgrouping, our study cohort comprises 2 (1.3%) very good, 68 (44.4%) good, 40 (26.1%) intermediate, 17 (11.1%) poor, and 26 (17.0%) very poor subgroups. We grouped mutations into larger sets or pathways and examined pattern of occurrence between these groups. Of the 153 patients, 132 (86%) contained at least one mutation in one of eleven categories defined to have biological function and have putative roles in MDS pathogenesis (Figure 1a and Figure 1b). Mutations were distributed over 153 patients, however no particular mutation seen from both patients in the very good prognostic subgroup. The most common mutations identified include genes involving: Histone modification - 26% good, 23% intermediate, 47% poor, and 15% very poor; DNA methylation - 50% good, 53% intermediate, 35% poor, and 23% very poor; Splicing factors - 54% good, 53% intermediate, 24% poor, and 19% very poor; Transcription factors - 38% good, 45% intermediate, 65% poor, and 31% very poor; Signal transduction pathways - 21% good, 35% intermediate, 35% poor, and 19% very poor; and Tumor suppressor TP53 - 6% good, 5% intermediate, 12% poor, and 85% very poor. Notably, emerged mutations from follow-up samples were seen in 6 (9%) patients in the good (primarily involving the cohesin complex, transcription factors, signaling, and DNA repair genes), 2 (5%) in the intermediate (involving DNA methylation and transcription factor genes), and 3(8%) in the very poor (involving DNA methylation and transcription factor genes) prognostic subgroups (p > 0.05). Within the good prognostic CN-MDS subgroup, 10% (6/58) of patients had emerged mutations from follow-up specimens, from which 1 (9%) patient transformed to AML, acquiring mutations involving multiple genes in various cellular pathways in the cohesin complex, signaling, transcription factor, and DNA repair mechanisms. We also noted that 2 or more co-mutations were identified in 69% (40/58) of CN-MDS, occurring in up to 11 genes in a single patient (Figure 2).
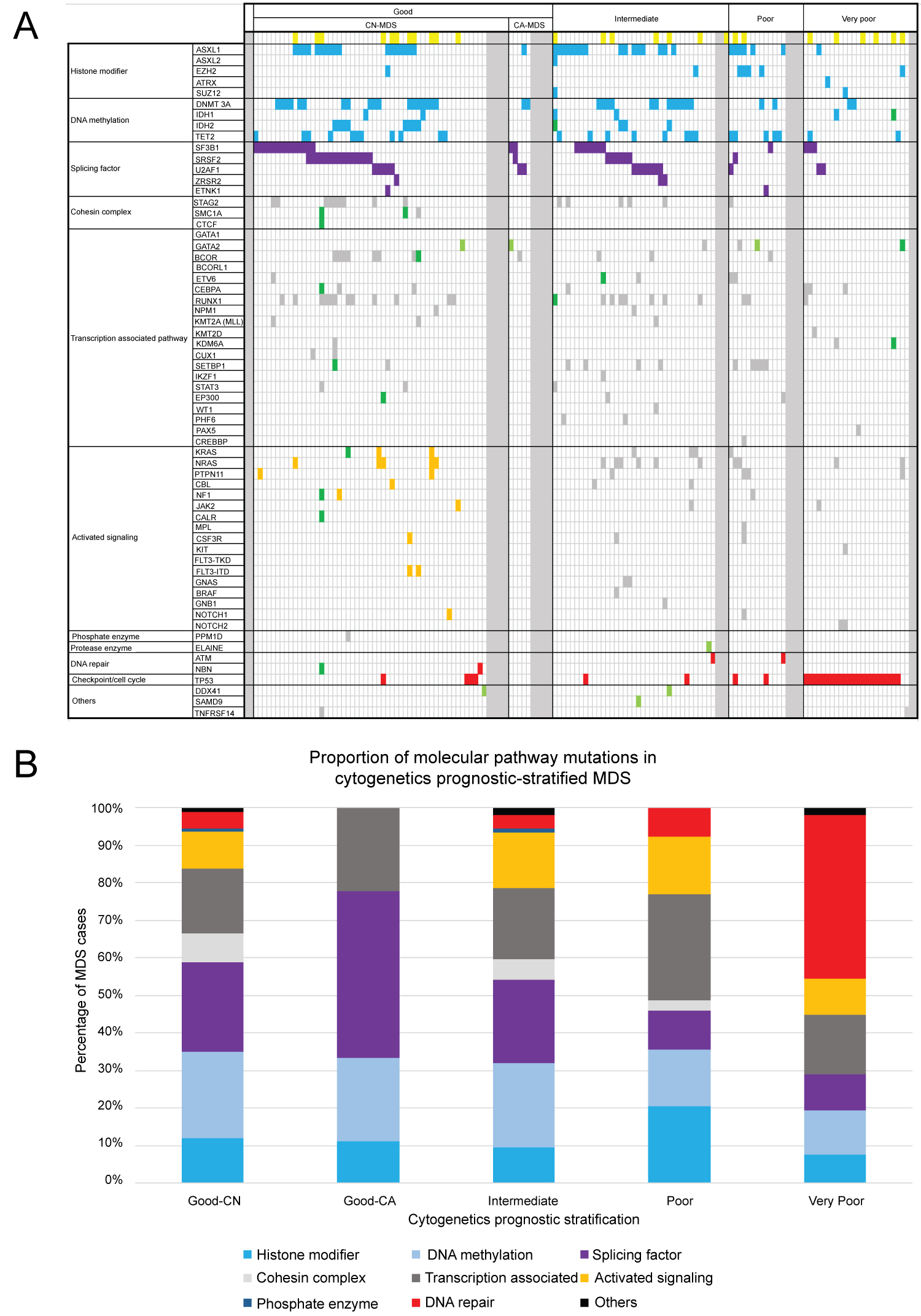 Figure 1 (a,b): Organization of mutations in pathways of associated genes. Illustrated are somatic nonsynonymous mutations in individual genes, grouped in eleven categories. Of the 153 patients, 132 (86%) contained at least one mutation in one of the genes listed. Yellow boxes correspond to patients initially diagnosed with MDS, and subsequently transformed to AML somewhere in the disease coarse; Blue boxes were mutations involving epigenetic modifiers; Purple boxes were mutations involving splicing genes; Orange boxes were mutations involving signaling genes at initial diagnosis in good CN-MDS subgroup; Red boxes represent mutations in DNA repair mechanisms and checkpoint pathway; Green boxes were the emerged mutations from follow-up specimen; Light green boxes were the germline mutations identified at diagnosis; Grey boxes represent all other mutations, including samples that has no mutant clone detected. View Figure 1
Figure 1 (a,b): Organization of mutations in pathways of associated genes. Illustrated are somatic nonsynonymous mutations in individual genes, grouped in eleven categories. Of the 153 patients, 132 (86%) contained at least one mutation in one of the genes listed. Yellow boxes correspond to patients initially diagnosed with MDS, and subsequently transformed to AML somewhere in the disease coarse; Blue boxes were mutations involving epigenetic modifiers; Purple boxes were mutations involving splicing genes; Orange boxes were mutations involving signaling genes at initial diagnosis in good CN-MDS subgroup; Red boxes represent mutations in DNA repair mechanisms and checkpoint pathway; Green boxes were the emerged mutations from follow-up specimen; Light green boxes were the germline mutations identified at diagnosis; Grey boxes represent all other mutations, including samples that has no mutant clone detected. View Figure 1
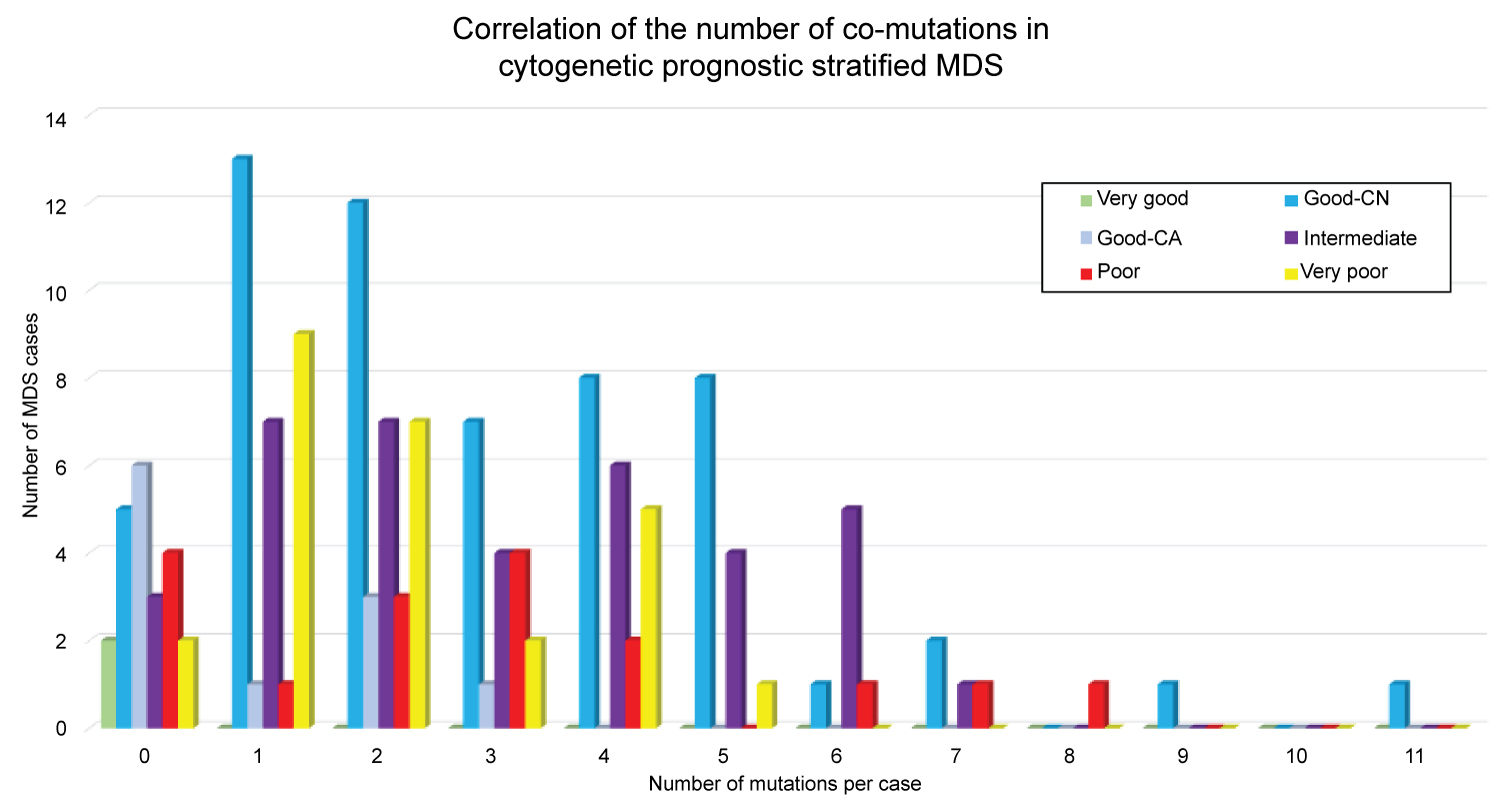 Figure 2: Correlation of the number of co-mutations in cytogenetic prognostic-stratified MDS. Illustration showing 2 or more mutations identified in 69% of CN-MDS (colored blue), occurring in up to 11 mutant genes in a single patient.
View Figure 2
Figure 2: Correlation of the number of co-mutations in cytogenetic prognostic-stratified MDS. Illustration showing 2 or more mutations identified in 69% of CN-MDS (colored blue), occurring in up to 11 mutant genes in a single patient.
View Figure 2
Mutations in genes involving mRNA splicing and DNA methylation predominate in the good and intermediate subgroups, while an increasing trend towards mutations in the transcription pathway genes were observed in the poor subgroup (comparing to the intermediate and good subgroups), however statistical significance was not reached (p = 0.2 and p = 0.06 respectively). Notably, our dataset shows that the presence of variant TP53 genes were not significantly different comparing intermediate and poor (p = 0.57) subgroups, but was significantly more common and prominently observed in the very poor prognostic subgroup (p = < 0.001).
Germline variants were also detected in a good number of cases. Most notably GATA2 gene was seen in 2 patients in the good and 1 in the poor subgroup, whereas germline DDX41 variant was identified in 1 patient in the good, and 1 in the intermediate subgroup. Other germline variants particularly SAMD9 and ELANE genes were identified in the intermediate subgroup (p > 0.05).
Within the good cytogenetics subgroup, we identified 58 (85%) patients with CN-MDS, 10 (15%) with CA-MDS, including 4 (6%) with sole del (20q), and 6 (9%) with isolated del (5q) at initial diagnosis. Notably, no particular mutation seen in 25% (1/4) of patients with sole del(20q) and 67% (4/6) of patients with isolated del(5q) (p = 0.52).
Somatic mutations in variety of genes were detected in 93% (53/58) of good CN-MDS prognostic subgroup. The common mutations in CN-MDS include genes involve in mRNA splicing (57%), DNA methylation (55%), transcription factors (41%), histone modifiers (29%), and signal transduction pathways (24%). Within this group, 11 (19%) patients progressed to overt s-AML.
Interestingly, time-dependent analysis showed that AML transformation was significantly higher in those patients who acquired mutations involving histone modifier (particularly ASXL1) and signaling genes (particularly NRAS, KRAS, PTPN11, CBL and NF1) at initial diagnosis (p = 0.008, hazard ratio 4.5 and p < 0.004, hazard ratio 8.9, respectively) (Figure 3). The median times to AML transformation were 39.3 and 22.2 months for mutant histone modifier and signaling genes, respectively. No significant differences to AML transformation observed among the other described pathways. The overall survival was similarly poor in CN-MDS carrying mutations of genes involving signaling pathways (p = 0.004, hazard ratio 9.1, median time to death of 39.3 months) compared to their wild-type counterparts. Multivariate logistic regression analysis further supports these findings showing that AML transformation is independently associated and significantly higher in those with mutations in histone modifier and signaling genes, with no significant factors contributing to age, sex, treatment protocol, and transplant history (Table 3).
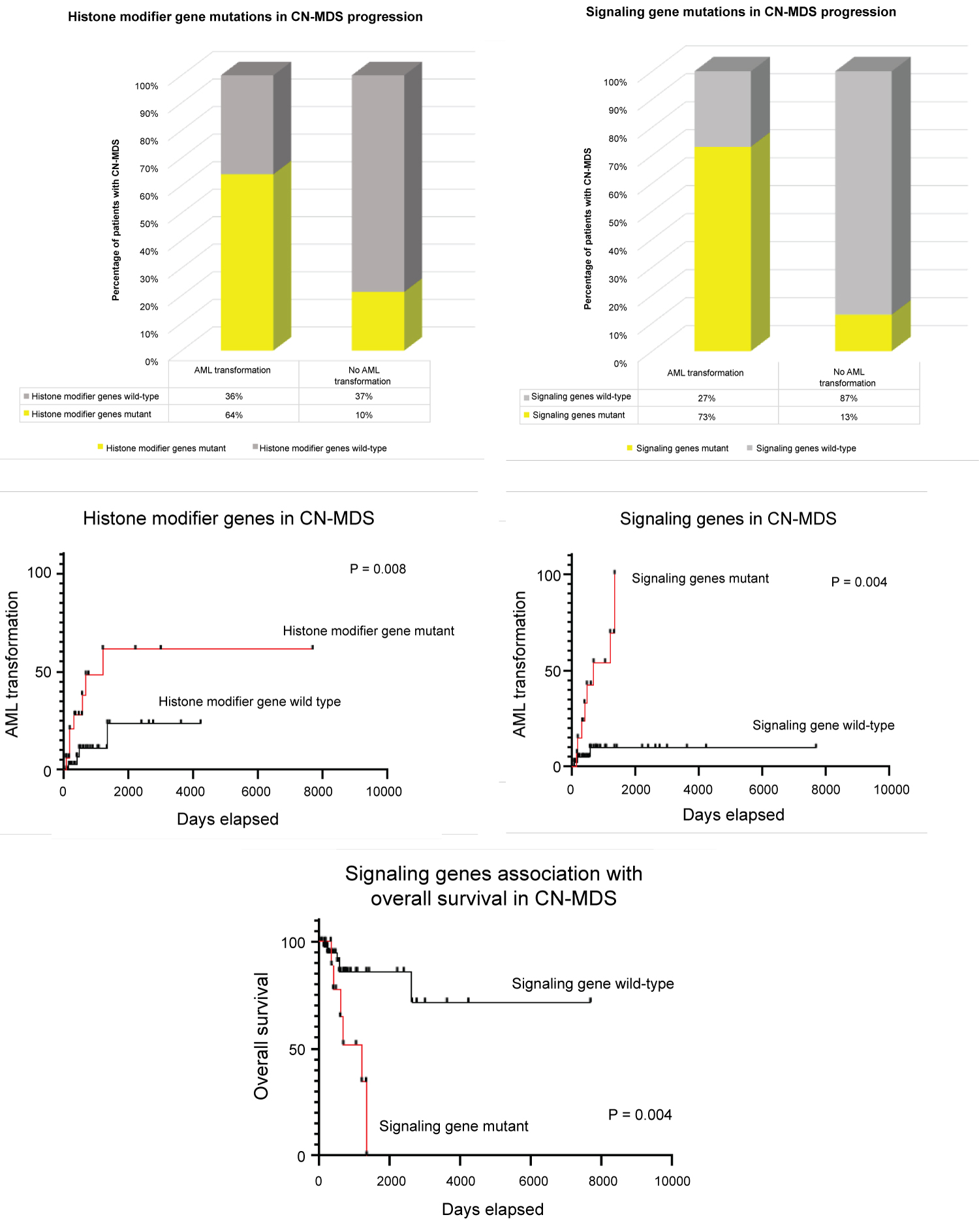 Figure 3: Significance of mutated histone modifier and signal transduction genes in good CN-MDS cytogenetics subgroup. (A) Shows 64% of patients with mutant histone modifier genes at initial diagnosis transform to AML, comparing to 10% of those who did not transform; (B) Shows 73% of patients with mutant signaling genes at initial diagnosis transform to AML, comparing to 13% of those who did not transform; (C and D) Time-dependent analysis showing AML transformation to be significantly higher in those patients who acquired mutations involving histone modifier and signaling genes at initial diagnosis (p = 0.008, hazard ratio 4.5 and p < 0.004, hazard ratio 8.9 respectively); (E) Poorer overall survival in those patients with mutant genes involving signaling pathways compared to their wild-type counterparts (p = 0.004). View Figure 3
Figure 3: Significance of mutated histone modifier and signal transduction genes in good CN-MDS cytogenetics subgroup. (A) Shows 64% of patients with mutant histone modifier genes at initial diagnosis transform to AML, comparing to 10% of those who did not transform; (B) Shows 73% of patients with mutant signaling genes at initial diagnosis transform to AML, comparing to 13% of those who did not transform; (C and D) Time-dependent analysis showing AML transformation to be significantly higher in those patients who acquired mutations involving histone modifier and signaling genes at initial diagnosis (p = 0.008, hazard ratio 4.5 and p < 0.004, hazard ratio 8.9 respectively); (E) Poorer overall survival in those patients with mutant genes involving signaling pathways compared to their wild-type counterparts (p = 0.004). View Figure 3
Our understanding about the genetics of MDS and related myeloid neoplasms has been dramatically improved over the past decade with the revolution of complex molecular testing such as sequencing studies, particularly NGS. By detecting large number of somatic mutations, an archive of driver and passenger mutant genes predisposing to MDS were discovered. The discovery of age-related clonal hematopoiesis also largely contributes to the understanding that not all myeloid-associated driver mutations can cause clinical, laboratory and microscopic alterations, suggesting a substantial overlap between myeloid neoplasms and age-related changes in healthy individuals. Reports show that approximately 10% of persons aged 70-80 carried one or more mutations indicative of clonal hematopoiesis [15]. Individuals with evidence of clonal hematopoiesis had 10-15 fold increased risk of developing hematologic malignancy, although not necessarily MDS [16,17]. The risk increased to 50 fold if the somatic mutation was present in 20% or more nucleated blood cells (corresponding to variant allele frequency (VAF) of ≥ 0.10 [15].
Molecular profiling studies on patient samples have suggested that somatic mutations are obtained in a step-wise fashion, where mutant genes with high VAF are proposed to occur early in disease development and mutations with lower VAF are thought to be acquired later in disease progression [16,18]. Although combinations of mutations have been functionally investigated, there remains uncertainty about the co-occurrence of somatic mutations and disease progression in MDS, particularly those that have normal cytogenetics.
MDS can be difficult to diagnose in a cytopenic patient, especially when blood and bone marrow morphological changes are equivocal, myeloblast percentage is not increased, and cytogenetics is normal. The WHO currently does not incorporate mutational findings as a concrete criterion in MDS diagnosis. However, in cases that meet morphologic criteria for MDS, typical somatic mutations strongly support the diagnosis [15]. We report the molecular spectrum of cytogenetic prognostic-stratified MDS from our 153 patients, and enumerated the most common genetic pathways involved in the pathogenesis of CN-MDS. NGS analysis revealed that 86% of MDS cases demonstrate somatic mutations using targeted panel of genes associated with hematologic malignancies. We showed that 91% of CN-MDS have underlying molecular alterations, which we believe are helpful diagnostic aid in confirming and providing an evidence of a clonal event in a patient with cytopenia, with equivocal morphologic and cytogenetic findings. We also demonstrated that the presence of mutations involving Histone modifier (particularly ASXL1) and signal transduction genes, (particularly NRAS, KRAS, PTPN11, CBL and NF1) to be strongly associated and detrimental events to AML transformation, and may contribute to survival information in patients with CN-MDS. Strikingly, it was identified that mutations of genes involving both histone/chromatin modification and signaling pathways occur in later in the disease biology of MDS, although these are not absolute rules, but show trends for the observed temporal acquisition [19].
Reports also showed that emergence of new driver mutations, even if they are still subclonal, can have significant implications for future disease course [19]. In this study, 10% of patients in the good prognostic CN-MDS subgroup had emerged mutations from follow-up specimens, from which 1 (9%) patient transformed to AML acquiring mutations involving multiple genes, within the cohesin complex, signal transduction, transcription factor, and DNA repair mechanism pathways. These findings may suggest that occurrence of mutations within these biological pathways may occur as late events in the pathogenesis and are detrimental to AML transformation.
In MDS, one of the strongest predictors of outcome is the number of driver mutations identified in a particular patient at initial diagnosis. Twenty percent (20%) of the mutations in patients with 1 driver mutation map to genes mutated in 2% of cases [19]. In our study, 2 or more occurring co-mutations were identified in 69% (40/58) of CN-MDS (occurring in up to 11 mutant genes in a single patient), indicating that increasing number of mutations detected at initial diagnosis maybe a strong supportive evidence of an underlying clonal process, and may predict a probable risk of AML transformation somewhere in the disease course. It was also recently proposed a new prognostic predictor for MDS based only on the variant status of 14 myeloid genes, which was shown to outperform the IPSS-R [20].
Somatic point mutations are previously reported to be common in MDS and are associated with specific clinical outcomes. Mutations involving variants of a histone modifier gene ASXL1 is one of the mutations previously identified and reported to be a significant predictor and independently associated with poor overall survival [21]. Similarly, frequent occurrence of signal transduction genes at initial diagnosis tends to have prognostic significance in the disease progression of MDS [22]. In our cohort, we demonstrate that significant proportion of CN-MDS patients with mutations involving histone modifier genes and signal transduction genes were significantly associated with AML transformation. This conclusion was consistent to previous report that signaling and histone modifying proteins are generally adequate for AML pathogenesis, and an activating mutation in a gene encoding a signaling protein might be a requirement for pathogenesis in adult de novo AML [23].
Complex cytogenetics in MDS is associated with mutant TP53 and can be associated with progression to AML [24]. In our data, TP53 mutant genes from patients with very complex cytogenetics (very poor subgroup) had paucity of mutations in other genes, suggesting that it may represent a distinct molecular subclass of MDS with a unique biological mechanism.
Our study provides an insight in the pathogenesis of myeloid transformation of CN-MDS and how clonal diversity contributes to AML transformation. We found out that detection of specific somatic mutations aid in predicting the clinical heterogeneity, and may help in predicting pathological behavior and prognosis of CN-MDS.
In conclusion, we demonstrate similarities and differences in the common mutational signatures and prognostic significance of molecular pathways in cytogenetic prognostic-stratified MDS. Mutations involving DNA methylation and RNA splicing factors were the most commonly mutated genes in the good and intermediate subgroups, with increasing mutations in the transcription factor pathway (65%) and signal transduction genes (35%) in the poor subgroup. Notably, mutations primarily involving the tumor suppressor gene TP53 (85%) were seen prominently in the very poor, but not in the poor subgroup. More importantly, presence of mutations in histone modifier genes and signaling genes were significant events to AML transformation in the good CN-MDS subgroup. Our results also suggest that in addition to conventional morphologic evaluation and cytogenetic findings, mutational analysis provide additional prognostic information regarding disease biology in CN-MDS cases and should be incorporated in the MDS diagnostic criteria as proof of an underlying clonal event, which may predict a particular clinical biological behavior and disease progression.
NGS often does not detect large deletions or duplications in other genes targeted on our panel. In addition, NGS does not detect mutations in the regulatory regions, deep introns, or highly homologous regions containing pseudogenes and/or highly repetitive regions. It is also intended to detect somatically-acquired variants in cancer-associated genes and was not intended to detect germline variants for the diagnosis of inherited cancer predisposition syndromes, although a probable finding of a germline mutations can be assumed based on VAF levels.