Activation and control of the immune system is regulated by costimulatory molecules as well as by checkpoint inhibitors. Checkpoints are essential in maintaining self-tolerance and minimizing collateral damage by modulating the immune response. Evasion of the immune system, one of the hallmarks of cancer, has been found to include interference with checkpoints by tumor cells as one of the evasive mechanisms. Tumor cells express molecules that when bound to their respective ligand or receptor, send out inhibitory signals that block T-cell activation. Specific antibodies have been engineered against these immunosuppresive molecules (mainly CTLA-4 and PD-1) such that the T-cells can exert cytotoxic anti-tumor effects. These antibody therapies have been found to be very effective for a number of malignancies, especially melanoma and lung cancer. The downside is that this therapy comes with serious adverse immune-mediated events, a direct consequence of releasing the brakes of the immune system. In comparison with patients with a deficiency in the Autoimmune Regulator (AIRE) transcription factor, the side effects however are manageable and certainly acceptable in light of the otherwise fatal underlying disease. Combination of different checkpoint inhibition antibodies, including novel immunoregulatory molecules may further enhance the effectiveness of this form of therapy and broaden the range of susceptible tumors. The vision of the hereafter for immunotherapy by checkpoint inhibition therefore looks bright.
AIRE deficiency, Cancer immunotherapy, Checkpoint inhibition, Immune-related adverse events, Immune response
The painting Visions of the Hereafter by Jheronimus Bosch shows in four panels that man can either go to hell or to heaven. The four panels (Fall of the Damned, Hell, Earthly Paradise, and Ascent into Heaven) were painted between 1510 and 1515 and are on permanent display in the Palazzo Grimani in Venice, Italy. On the occasion of the 600th anniversary of the death of Jheronimus Bosch, the paintings were returned to Hertogenbosch in 2016 for the exhibition in the town where he has lived all his life. In contrast to most of Bosch's other work, the meaning of these four panels is straightforward: if you live a good life you will go to heaven, if not, the devils will get you and you will go to hell. In the Fall of the Damned panel (Figure 1) an apparent helpless man is attacked by a devil. It can also be viewed, although almost certainly that was not the intention of Bosch, as a defenseless human body being attacked by a tumor. This indeed was the prevalent paradigm until recently: humans with an inadequate defense system (defenseless) are vulnerable for tumors.
The immune system has evolved to protect against invasion by microbiological dangers and to maintain the body's internal environment. At birth certain defense mechanisms, particularly of the innate immune system, have already developed. The innate immune system recognizes molecular patterns of micro-organisms and subsequently kills and destroys them via a variety of effector mechanisms. However, as a newborn grows older, the previously naïve defense mechanisms mature and become more specific and stronger. They have adapted to previously encountered pathogens in order to develop attacks specific to those pathogens (immunological memory). The acquired or adaptive immune system is based on specific recognition of antigens on micro-organisms by antigen receptors expressed on B- and T-lymphocytes, the major players of the adaptive immune system. Precursors of both T-lymphocytes (T-cells) and B-lymphocytes (B-cells) are generated in the bone marrow, however T-cells undergo 'education' in the thymus and B-cells predominantly mature in the bone marrow, hence their names. They reside in the peripheral lymphoid tissue and can travel to the site of infection through the bloodstream. Before they can participate in the immune response, B- and T-cells need to be activated in order to become effector cells that recognize and respond to antigen.
T-cell activation can lead to the differentiation into three different effector T-cells: Cytotoxic T-Cells (CTL's), helper T-cells (TH-cells) and regulatory T-cells (Tregs). CTL's kills cells that have been infected by a pathogen with very effective destructive mechanisms. TH-cells help activating several other immune cells such as B-cells, macrophages and CTL's by secreting cytokines (intercellular chemical messengers) and expressing co-stimulatory molecules. A cytokine especially important for the differentiation and proliferation of effector T-cells is Interleukin-2 (IL-2). Tregs, as their name suggests, regulate the immune response so that the reaction is controlled and the collateral damage, damage to healthy tissue as a result of attacking the pathogen, is minimalized.
T-cell activation starts with recognition of an antigen processed and presented by an Antigen-Presenting Cell (APC) in the Major Histocompatibility Complex (MHC) molecule. The TCR binds to the antigen part of the complex and either CD4 or CD8 molecules present on the T-cell surface act as co-receptors and bind to the MHC-part of the complex. If the T-cell bears a CD4 co-receptor, then the cell is referred to as a CD4+ T-cell and will interact with MHC-class II-antigen complexes. CD8+ T-cells, T-cells that express CD8 molecules, bind to MHC-class I-antigen complexes. Whether a T-cell is CD4+ or CD8+ is the reason why the MHC class determines what effector cell the T-cell will differentiate into. When the MHC-antigen complex binds to a CD4+ T-cell, it will activate the cell to become a TH-cell. The T-cell will be activated to differentiate into a CTL when it is CD8+ [1]. The interaction between MHC-antigen complexes and TCR and CD4 or CD8 receptors is referred to as the 'first' signal of T-cell activation.
Successful activation of T-cells requires co-stimulation, which is provided by the interaction of surface molecules on T-cells and their counter structures on the APC. The main co-stimulatory signal, the 'second' signal of T-cell activation, is the binding of the CD28 molecule on the T-cell surface with a B7.1 or B7.2 molecule (also known as CD80 and CD86 respectively) expressed on the APC. In the absence of a second signal, the T-cell will go into a state of anergy, functional inactivity.
Because the immune system is extremely effective at eliminating potentially harmful substances from the body, and therefore potentially destructive, it should be kept tightly under control. Any immune response inevitably leads to collateral damage and temporary changes of the physiology of major organ systems, which requires anergy from the body to repair. It thus would be very dangerous if the immune system would respond to every trigger and substance that may enter or already resides within the body, because not all of these substances are harmful. What especially should be avoided is an immune response against body-own components (autoantigens). Normally, the immune system is tolerant to body-own components: self-tolerance. Self-tolerance is induced during the development of B- and T-cells in bone marrow and thymus, respectively (central tolerance). Self-reactive cells that may escape these selection processes are kept under control in the periphery by regulatory T-cells (Tregs): peripheral tolerance. These inhibitory (suppressive) mechanisms can be considered the 'brakes' of the immune system. Binding of co-stimulatory molecules, or lack thereof, is often referred to as immune checkpoints, because they keep the immune response 'in check' and they should be passed only in an immune response to a pathogen [2]. These checkpoints are necessary to maintain self-tolerance and limit collateral damage [2-4].
 Figure 1: Fall of the Damned, a panel of Visions of the Hereafter (1505-1515) by Jheronimus Bosch (Palazzo Grimani, Venice, Italy) (http://boschproject.org/bosch_in_venice.html). View Figure 1
Figure 1: Fall of the Damned, a panel of Visions of the Hereafter (1505-1515) by Jheronimus Bosch (Palazzo Grimani, Venice, Italy) (http://boschproject.org/bosch_in_venice.html). View Figure 1
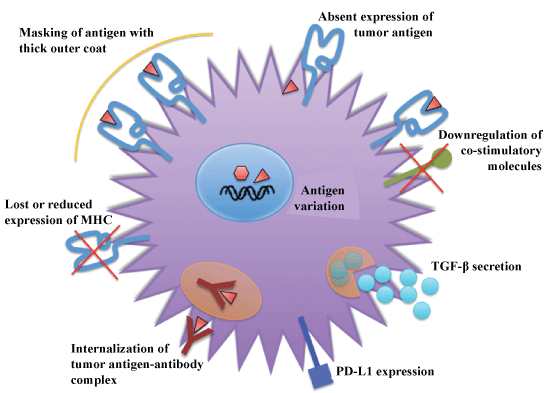 Figure 2: Mechanisms of evasion of immune system by tumor cells. View Figure 2
Figure 2: Mechanisms of evasion of immune system by tumor cells. View Figure 2
Cancer is the result of many genetic and epigenetic changes that occur as a consequence of genetic damage (mutations) caused by environmental agents or inheritance. These alterations lead to expression of altered protein products that the cancer cells subsequently also express on their surfaces. In most cases, the immune system recognizes these proteins as 'non- or altered-self' and eliminates these cells. Rarely, cancer cells can escape the immune system. The importance of immune surveillance for protection against cancer is underscored in patients with immunodeficiencies who have a higher incidence of tumors. One hallmark of cancer therefore, is evasion of the immune system, as well as self-sufficiency in growth signals, evasion of cell death, limitless replicative potential, and induction of angiogenesis. Several mechanisms that cancer cells use to evade the immune system have been identified and include the elimination of immunogenic cancer cells, loss or reduced expression of MHC molecules to escape attack by CTL's, masking of antigen with a thick outer coat, immune suppression through production of Transforming Growth Factor-β (TGF-β), internalization of tumor antigen-antibody complex, antigen variation, absent expression of tumor antigen, and down regulation of co-stimulatory of molecules (Figure 2).
Although cancer cells have the ability to escape elimination by the immune system, they also need the right circumstances within the tissue they originate in order to proliferate, grow, invade, and metastasize. These local circumstances in which the cancer cells develop (the tumor microenvironment) include the cells (e.g. immune cells, signaling molecules, extracellular matrix, and blood vessels that surround and feed the tumor [5].
Many different treatments against cancer have been developed over the years; however the most prevalent conventional therapies remain surgery, chemotherapy, and radiation. For chemotherapy and radiation, which mostly target (rapidly) dividing cells, a balance should be found between eradicating the cancer cells and sparing healthy tissues. As for surgical resection of the cancer, the tumor microenvironment should be taken into account, because it may stimulate any remaining cancer cells if not resected. Since the rise of immunotherapy, which refers to enhancement or suppression of the immune system as a therapy, in the mid twentieth century, many (clinical) researchers have attempted to replace the conventional, quite invasive, treatments with new immunotherapies.
An example of first generation cancer immunotherapy is the dendritic cell vaccine approach [6]. Another example is the opsonization, or labeling of the tumor cells with specific antibodies [7]. Both of these examples of first generation cancer immunotherapy focus on improving the immune system of the patient by enhancement of antigen presentation in order to induce a T-cell-mediated immune response against the cancer cells.
Recent research has shown that apart from antigen presentation which can be insufficient to induce adequate anti-tumor responses, the behavior of the tumor also influences the activation of the T-cells, as will be discussed below [8]. The previously mentioned immune checkpoints seem not to be binding to a stimulatory ligand (like B7.1) as in a normally occurring situation, but rather the T-cell activation is held up by the checkpoint because an inhibitory ligand binds to the co-stimulatory receptor molecule instead. Tumors often take advantage of this already existing mechanism (checkpoints are normally also necessary in regulating an immune response) by expressing the inhibitory ligands that can bind to the co-stimulatory receptors on T-cells. See Figure 3 for a selection of the many co-stimulatory receptors and their ligands [2]. In a new cancer immunotherapy, a specifically engineered antibody blocks the inhibitory ligand so that it can no longer bind to the checkpoint. This type of therapy is known as checkpoint inhibition.
Creating specific antibodies that block these ligands stops the tumor from inhibiting the immune response, but also takes away the regulatory function. In other words, the safety brakes of the immune system that normally keep the immune response under control are now released. One can imagine that this can create serious complications or side effects of the treatment in other parts of the body, where inhibition of the checkpoints is necessary because again, they are crucial for maintaining self-tolerance and modulating the duration and amplitude of the immune in order to minimize collateral tissue damage [2].
This paper will investigate checkpoint inhibition as an immunotherapy for cancer with regard to the extent to which the brakes of the immune system can be released, without causing serious side effects. A fine balance between sufficient activation against tumor cells and over activation against (non-transformed) cells and molecules of the body should be achieved if checkpoint inhibition were to be the ideal novel cancer treatment.
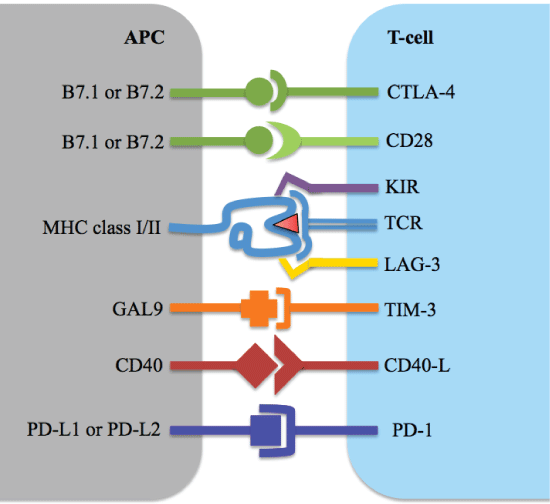 Figure 3: Co-stimulatory receptors and their ligands on antigen presenting cells and T-cells. View Figure 3
Figure 3: Co-stimulatory receptors and their ligands on antigen presenting cells and T-cells. View Figure 3
The finding that tumors often express the receptors for or ligands of inhibitory pathways involving checkpoints for an immune response has sparked novel forms of immunotherapy of cancer. Two major inhibitory molecules of the immune response are the Cytotoxic T-Lymphocyte-Associated Antigen 4 (CTLA-4) and Programmed Cell Death Protein 1 (PD-1) [2-4,7,9,10]. The antibodies against these molecules which are currently being used in clinical studies are indicated in Table 1.
CTLA-4 (Cytotoxic T-Lymphocyte-Associated Protein 4) can also bind to B7.1 and B7.2. In fact, CTLA-4 has a much higher affinity for the B7 ligands than CD28, thereby able to out-compete the CD28 receptor [11]. When CTLA-4 binds to B7, it can no longer interact with CD28 and the second signal is missing. Since the B7 ligands are expressed on APC's, the CTLA-4-mediated immune regulation mostly resides in secondary lymphoid organs where T-cell activation takes place [4]. Normally, CTLA-4 is expressed on activated T-cells (both TH-cells and CTL's) and Tregs to regulate the amplitude of T-cell activation [2-4]. More specifically, regulation occurs through down modulation of TH-cells (which have immunostimulatory effects) and stimulation of the immunosuppressive activity of Tregs [12]. CTLA-4 also interacts with the so-called 'TCR stop-signal', which is a signal to ensure that an activated T-cell will have sufficient time to interact with the MHC-antigen complex [13]. The importance of this function is dramatically demonstrated in Ctla4-knockout mice, which have an overactive immune system resulting in extreme lethal autoimmune reactions [14].
Table 1: Checkpoint molecules and their specific antibodies. View Table 1
Table 2: Autoimmune toxicity. View Table 2
Since CTLA-4 has an inhibitory effect on the immune system, which the tumor cells seem to be taking advantage of in order to escape it, CTLA-4 has become a target for immunotherapy against cancer. In order to inhibit the activity of this CTLA-4 so that the ligand itself cannot block the checkpoint (thereby inhibiting T-cell activation), specific anti-CTLA-4 antibodies have been engineered. After it was demonstrated that antibodies partially blocking CTLA-4 indeed lead to improved antitumor immunity in mice [15], anti-CTLA-4 therapy was introduced in clinical trials. In 2003 Hodi, et al. published the first clinical report of CTLA-4 antibody treatment in cancer [16]. The study started in 2000 and involved nine patients, seven metastatic melanoma (skin cancer) and two ovarian carcinoma patients. These patients were previously vaccinated and injection with the CTLA-4 blocking antibodies stimulated tumor necrosis (cell death) in five out of nine patients [16]. Although the study did show some antitumor immunity induced by the anti-CTLA-4 antibodies, Phan, et al. were the first to clearly demonstrate that CTLA-4 blockage induced tumor regression in three out of the fourteen treated melanoma patients. Important to mention is that out of the fourteen patients treated, six developed grade III to IV autoimmune toxicities [17]. Adverse events like autoimmune reactions or toxicities are graded on a scale from 0 to V (Table 2 adapted from reference [18]).
A breakthrough was achieved by a phase III randomized double-blind clinical trial conducted by the investigators of the Bristol-Myers Squibb pharmaceutical company [19]. This lead in 2011 to the FDA approval of this antibody now called ipilimumab [20]. In this study, a group of 676 metastatic melanoma patients were randomly assigned in a 3:1:1 ratio to receive either ipilimumab plus a glycoprotein 100 (gp100) vaccine containing the melanoma antigen peptide, ipilimumab alone, or gp100 alone. The median overall survival was 10.0 months in the ipilimumab-plus-gp100 group and 10.1 months in the ipilimumab-alone group compared to 6.4 months for the gp100-alone group. Overall survival rates at 24 months were 21.6, 23.6 and 13.7% in the ipilimumab-plus-gp100 group, the ipilimumab-alone group, and the gp100-alone group, respectively. The ipilimumab-alone group had the highest percentage of patients with an objective response or stable disease. Approximately 60% of the patients treated with ipilimumab had immune-related adverse events. Grade III or IV adverse effects were 10-15% in the ipilimumab groups compared to the 3% in the control group. More importantly, 50% of the study drug related deaths (in 2.1% of the patients) were associated with immune-related adverse events [21]. This shows that although ipilimumab seems to be more effective as compared to tumor vaccination, it is also the treatment with the highest frequency of immune-related adverse effects.
In a follow-up study, two years after treatment in the original clinical trial, 83% of surviving patients (20% of initial participants) were treated with ipilimumab. Among these patients, 14% had high-grade immune-related adverse events [22]. Although the immune toxicities are considerable, they balance the anti-tumor reactions, which lead to an overall survival of more than two years for nearly 20% of the melanoma patients.
For other tumors, at best a partial response is obtained in a minority of patients. The immune-mediated side effects are severe.
Anti-CTLA-4 therapy has been investigated in many cancers other than melanoma with mixed results from effect in combination with other therapies to no effects. Ipilimumab and tremelimumab have been investigated in lung (both small-cell and non-small-cell), pancreatic, hepatic, gastroesophageal, head and neck, breast, mesothelioma, and glioblastoma tumors [23,24]. Several studies demonstrate a trend towards improved overall survival in patients with both small-cell and non-small-cell lung cancer treated with ipilimumab in combination with chemotherapy [25-27], which warrants further investigation for ipilimumab as a first-line treatment [22]. No effect, besides many adverse events [28], was seen in pancreatic adenocarcinoma [29] and only partial response in the minority of the patients with hepatocellular carcinoma [28] or gastroesophageal cancer [30] treated with ipilimumab or tremelimumab (another anti-CTLA-4 antibody (Table 1), respectively.
The current literature demonstrates that anti-CTLA-4 therapy is being investigated in many types of cancers, but has only really shown to be of clinical significance in metastatic melanoma and possibly in lung cancer; two diseases in which improved survival rates outweigh the frequent moderate to severe immune toxicities.
The second checkpoint inhibitor, PD-1 (Programmed Death Protein 1) is especially active in the tumor microenvironment and interacts with PD-1-ligand 1 (PDL1), the predominant ligand, and PD-1-ligand 2 (PD-L2). PD-1 is expressed on the membranes of activated T-cells, Tregs and also on B-cells and Natural Killer (NK) cells [2]. PD-L1 is highly expressed in various cancer types such as melanoma, non-small cell lung cancer, glioblastoma, multiple myeloma, and many more, and upregulated on tumor cells through activation of key oncogenic pathways like the Mitogen-Activated Protein Kinase (MAPK) pathway and by cytokine stimulation, especially by Interferon-Gamma (IFNγ) produced in the tumor microenvironment [31]. Binding of PD-1 with PD-L1 inhibits TCR-mediated lymphocyte proliferation and cytokine secretion, but seems to have a bigger effect on inhibiting levels of CD28-mediated costimulation [32]. It has now become clear that CD28 is the primary target for PD-1 signaling, being inactivated through PD-1-recruted Shp2 phosphatase [33]. PD-L2 is expressed primarily on APCs and has a much more restricted tissue and tumor type distribution compared to PD-L1 [31]. The inhibitory signal elicited by PD-1 is to limit T-cell activity in an inflammatory reaction and to protect against autoimmunity within the tissues. Tumor cells seem to take advantage of this regulation and express PD-1 ligands to circumvent T-cell-mediated destruction [3]. In animal models, the phenotype of PD-1-/- mice is much less severe than CTLA-4 deficient mice. PD-1-/- mice develop late-onset lupus-like proliferative glomerulonephritis and arthritis [34].
Apart from the fact that PD-1 expression is broader than CTLA-4 expression (which is limited to T-cells), it also relevant to consider which immunological factors induce the expression of the PD-1 ligands on the tumor cells. By expressing PD-1 ligands the tumor cell becomes resistant to both innate immune attack [35] as well as adaptive immune attack [4]. Tumor Infiltrating Lymphocytes (TILs) secrete IFNγ [3], which is used by the tumor cell to up regulate its PD-1 ligand expression [36].
Because PD-1 is also expressed on Tregs, B-cells and NK cells, anti-PD-1 directed immunotherapy might limit immunosuppression by Tregs, enhance NK cell activity and may enhance antibody production as well [2]. This is surprising, since the major response is thought to be minimization of the inhibitory signals towards activated TILs within the tumor microenvironment. Increased PD-1 ligand expression for longer periods of time, such as in chronic inflammation and cancer, can also lead to anergy of antigen-specific T-cells. Anti-PD-1 may partially reverse this process and reactivate some of these previously anergic T-cells [2,9,10]. The emerging picture is that PD-1 blockade is most effective at time of T-cell activation and less so in the so-called exhausted T-cells. This may limit widespread unnecessary application of checkpoint inhibition, especially in established tumors [37,38].
Several clinical trials have investigated the efficacy and safety of anti-PD-1 therapy and specific anti-PD-1 antibodies such as nivolumab, pembrolizumab, and pidilizumab have been developed (Table 1). Nivolumab was approved for metastatic melanoma in 2014 and for metastatic squamous Non-Small Cell Lung Cancer (NSCLC) and metastatic renal cell carcinoma in 2015 [39,40]. The approval of nivolumab for metastatic melanoma was based on a phase III clinical trial in which efficacy and safety of nivolumab was compared with chemotherapy. Thirty-two percent of the patients in the nivolumab group showed objective responses versus 11% in the chemotherapy group. Treatment-related adverse events developed in the majority of patients, but grade III or IV in only 9% [41]. The approval of nivolumab as a treatment for NSCLC was based on two clinical trials. The first was a single-arm trial with 117 patients who received nivolumab after two or more previous treatments, and 13% of them had objective responses. Immune-mediated adverse events included mostly skin and gastrointestinal symptoms and occurred in only 10% of the patients [42]. The second, international trial compared nivolumab with docetaxel (standard chemotherapy). The overall survival, response rate and progression-free survival were all significantly better with nivolumab. Also the risk of death was lower, as well as the number of adverse events [43].
For metastatic renal cell carcinoma, nivolumab was studied in patients who did not respond to prior anti-angiogenic therapy. As compared to everolimus, median overall survive was nearly half a year longer with nivolumab, with less adverse events [44].
Nivolumab is not effective as treatment for colorectal and prostate cancer [45-47]. However, Le, et al. found that a subset of patients with colorectal cancer (i.e. those with a mismatch-repair deficiency) did respond to pembrolizumab, another anti-PD-1 antibody [48-50]. Grade III or IV adverse events occurred in 41% of the patients [50]. In patients with ovarian cancer, 24% showed partial response or stable disease. No responses were observed in patients with pancreatic, gastric or breast cancer. Information on adverse events within the ovarian carcinoma patient group is not specified. In the total group of patients, 61% developed treatment-related adverse events, of which 9% experienced grade III or IV adverse events [51].
As indicated above, both anti-CTLA-4 and anti-PD-1 therapies can have impressive clinical results, but which of these two would be preferred? The efficacy and safety of pembrolizumab (anti-PD-1) versus ipilimumab (anti-CTLA-4) in patients with advanced melanoma was investigated by Robert, et al. [52]. In this study, 6-month progression free survival rates for pembrolizumab (47%) were significantly higher as compared to ipilimumab (27%). Treatment-related adverse events ranging from grade III to V occurred in 13% of pembroluzimab treated patients and 20% in the ipilimumab group, one death was considered treatment-related. In a meta-analysis conducted by Barbee, et al., a PubMed search was performed and clinical trials assessing the safety and efficacy of ipilimumab, nivolumab, and pembrolizumab in cancer were selected [53]. They found that ipilimumab and nivolumab are most recommendable for treatment of metastatic melanoma based on survival rates. However, nivolumab and pembrolizumab are considered safer compared to ipilimumab. Generally, immune-related adverse events are less common and less severe in patients treated with anti-PD-1 therapy. Long-term follow-up results of anti-PD-1 therapy have not been published because of the more recent approval, so durability of the treatment cannot be compared yet to anti-CTLA-4 therapy.
Furthermore, as indicated above, anti-CTLA-4 appears to be effective as a treatment only for metastatic melanoma and NSCLC, whereas anti-PD-1 therapy has also shown improvement in other tumors [54].
Because anti-CTLA-4 and anti-PD-1 target different pathways, combination could have a synergistic effect. In a preclinical melanoma mouse model, dual blockade of PD-1 and CTLA-4 indeed acts synergistically [55]. Dual checkpoint inhibition of PD-1 and CTLA-4 has shown an overall survival benefit in a greater proportion of patients with advanced melanoma compared to either ipilimumab or nivolumab treatment alone [56]. Objective response rates were also better in combination therapy [21,54]. Combination of anti-CTLA4 and anti-PD-1 seems to be more effective, likely because they synergistically enhance co-stimulation of T-cells through CD28. For other diseases, especially lung cancer, combination checkpoint therapy is currently investigated [57-60].
Checkpoint inhibitors have been investigated in combination with other cancer treatments such as chemotherapy, targeted therapy, radiotherapy and other forms of immunotherapy [61]. As reviewed by Antonia, et al. especially ipilimumab has shown positive results in combination with chemotherapy for melanoma and lung cancer [62], but also positive effects were demonstrated in combination with radiotherapy in stage IV metastatic melanoma patients [63]. When a group of 22 patients underwent radiotherapy and were several days later treated with 4 cycles of ipilimumab, in 18% of them a partial response was observed, while another 18% remained stable. However, the remaining 64% of these patients had no benefits of the combination therapy. In another study with also 22 patients, 50% showed an improving clinical status, including complete responses [64]. In this study 4 cycles of ipilimumab treatment preceded radiotherapy by 5 days. Ipilimumab in combination with targeted therapy resulted in objective response rates in patients with renal cell carcinoma. Besides chemotherapy, ipilimumab has been evaluated in combination with radiotherapy, which has also shown benefit for patients with melanoma and prostate cancer. Combination of checkpoint inhibitors with other immunotherapies such as cytokine therapy and inhibition of IDO (indoleamine 2,3-dioxygenase, an immunosuppressive enzyme) are currently under investigation and may very well be promising [62]. Lastly, for an extensive overview of clinical examples and their significant outcomes see also the review by Sindoni, et al. [65].
Currently in phase II, III, and even phase IV trials, are the checkpoint inhibitors against PD-L1. Atezolizumab, durvalumab, and avelumab (Table 1) are under investigation for several types of cancers and have even been FDA approved for certain late stage cancers. The results of the OAK study and the POPLAR study, both published in the Lancet, resulted in the FDA approval of atezolizumab in 2016 for the treatment of patients with non-small cell lung cancer whose disease had progressed after or during chemotherapy [66]. In the OAK study, 1225 patients were enrolled and they were randomized to atezolizumab or docetaxel. Longer overall survival and fewer adverse events were observed in the atezolizumab group [67]. In the POPLAR study, 287 patients were randomized to atezolizumab or docetaxel and longer overall survival and fewer adverse events were also found in the atezolizumab group in this study [68]. Atezolizumab is now being studied in a phase IV clinical trial for the treatment of metastatic urothelial carcinoma [69]. In phase II and III clinical trials, atezolizumab is studied as treatment for ovarian, fallopian, peritoneal, breast, lung, bladder, and renal cancers and also in melanomas [70-75].
Durvalumab received accelerated FDA approval in 2017 for the treatment of locally advanced or metastatic urothelial carcinoma after failed chemotherapy [76]. The approval was based on a phase I/II trial in which the response rate of all patients was 17% and in patients with high PD-L1 expressed tumors 26.3%. Grade III and IV adverse events occurred in 43% of the patients of which immune-related adverse events were pneumonitis, hepatitis, colitis, thyroid disease, adrenal insufficiency [76]. Durvalumab is currently studied in phase I and II trials in prostate, oropharynx, pancreatic, renal, esophageal, breast, and colorectal cancers, often in combination therapy with tremelimumab [77] and phase III trials in non-small cell lung cancer [78]. Avelumab is being investigated in the same type of cancers, also in phase I, II, and III clinical trials [79,80].
As indicated above, checkpoint inhibition, specifically PD-1 and CTLA-4 inhibition, gives favorable results but with undesirable side effects. In order to evaluate to what extent the immune-related adverse events should be taken into account when considering anti-PD-1 and/or anti-CTLA-4 therapy, the adverse events are compared to the symptoms of the Autoimmune Regulator Transcription Factor (AIRE) deficiency. When T-cells mature in the thymus, Tissue-Restricted Antigens (TRA) are presented to induce central immune tolerance to self. Gene expression of TRAs is partly controlled by the AIRE gene and patients deficient in this gene develop a range of severe autoimmune diseases [81]. AIRE deficient patients develop Autoimmune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy (APECED) syndrome, which is diagnosed when the patient shows two of the three following symptoms: chronic mucocutaneous candidiasis, hypoparathyroidism, or Addison's disease [82]. Other conditions such as hepatitis, pernicious anemia, fatigue, chronic diarrhea, and vitiligo have also been described [83].
The most common immune-related adverse events reported with anti-PD-1 or anti-CTLA-4 therapy include dermatologic toxicity, diarrhea/colitis, hepatotoxicity, and endocrinopathies, although other systems such as the respiratory system may also be affected [84]. More specifically, the most commonly observed symptoms include pruritus, rash, vitiligo, diarrhea, colitis, hypo- or hyperthyroidism, hypophysitis, fatigue, increased aminotransferases, and pneumonitis. Also an immune-mediated thrombocytopenia has been reported as a side effect [85]. A comparison of the symptoms of AIRE deficient patients and the immune-related adverse events with checkpoint inhibition based on the previously mentioned clinical trials and several additional sources [82-84,86,87] is shown in Table 3.
Of the symptoms indicated in Table 3, 8 out of the 18 are manifested in both patients with APECED and in patients receiving checkpoint inhibition treatment. Kluger, et al. conducted a study evaluating the quality of life in APECED patients and found that depressive symptoms affected 29% of the patients [88]. General health, emotional well-being, and vitality were the most affected aspects of quality of life. As compared to APECED, there are fewer and less severe immune-related adverse events in checkpoint inhibition. Most adverse events are relatively easily manageable and involve the administration of topical or systemic glucocorticosteroids [84].
Considering the clinical success of anti-PD-1 and anti-CLTA-4 therapy in previously virtually untreatable cancers like metastatic melanoma and NSCLC, the supplementary consequences are acceptable, especially in comparison with AIRE deficient patients, who clearly suffer from more numerous and severe autoimmune conditions.
Table 3: AIRE deficiency symptoms compared to checkpoint inhibition immune-related adverse events. View Table 3
Note: X- Indicates that the symptom is present.
Since the clinical success of anti-CTLA-4 and anti-PD-1 therapy, these and other checkpoints are under the investigation as potential anti-tumor activity inducers. The clinicaltrials.gov registry lists 39 ongoing trials on checkpoint inhibition (https://clinicaltrials.gov/; accessed on July 5, 2017). Lymphocyte Activation Gene 3 (LAG-3) is expressed on activated T-cells, NK cells, B-cells, and dendritic cells and binds to MHC-class II molecule. Studies have shown that it is selectively upregulated on Tregs, which when blocked inhibits the immunosuppressive activity of Tregs. It has also been shown to enhance CTL activity in the absence of CD4+ T-cells, demonstrating that it also plays a role in CTLs [4].
T-Cell Membrane Protein 3 (TIM-3) blockade also shows potential as a cancer treatment. It has been shown to be upregulated in certain cancers, such as breast cancer, and inhibits TH1-cells. TIM-3 is nearly universally co-expressed with PD-1 on the majority of TILs and anti-TIM-3 antibodies enhance anti-tumor immunity [2,4].
Killer Inhibitory Receptors (KIRs), regulators of NK cell cytotoxicity, are also a target for immunotherapy. Many of these receptors are specific for the MHC molecules and also have inhibitory effects on T-cells and APCs, besides on NK cells. Antibody blockade of these KIRs results in activation of NK cells, inducing potent anti-tumor immune responses [2,4].
The elucidation of immune checkpoints and the involved pathways has provided new options for previously untreatable cancers. The body's natural immunological mechanisms can now be manipulated to induce effective anti-tumor immunity. Easily administrable checkpoint inhibitors have had astonishing effects in metastatic melanoma and NSCLC. In the last five years, several different anti-checkpoint antibodies have been developed, approved, and implemented. Although the immune-mediated negative side effects can be considerable, the clinically significant results of progression-free survival, overall survival, and objective response rates by far outweigh those side effects. When compared to the clinical spectrum of AIRE deficient patients, patients with a substantially impaired quality of life, the adverse events of checkpoint inhibition are acceptable and relatively easy to treat.
It should be emphasized that the success of checkpoint inhibition therapy still is based on the quantification of the responses measured on the Response Evaluation Criteria in Solid Tumors (RECIST) and Immune-Related Response Criteria (ir-RC) scales. These scales would not have to be used if the tumor would be completely eradicated [28].
Both the existing and upcoming therapies including other checkpoint inhibitors and genetically engineered anti-tumor T-cells may have promising clinical benefits. Future therapies may include strategies such as the development of specific antibodies that do not only target the evasiveness of the cancer, but also the other hallmarks of cancer, for instance self-sufficiency in growth signals and evasion of cell death. As for current anti-cancer therapy through checkpoint inhibition, adverse events may be severe. Indeed, recent meta-analysis indicates that fatal adverse events as well as immune-related adverse events are significantly higher in patients treated with immune checkpoint inhibitors [89-91]. However, the immune-related adverse events are better treatable than the previously untreatable underlying disease. The discovery and elucidation of current and potential future targets allows for a step forward in the fight against cancer. The vision of the hereafter therefore looks bright.