Single Nucleotide Polymorphisms (SNPs) in genes influencing hypercytokinemia have been implicated in the clinical manifestations of severe influenza infection. Genotype-phenotype studies in healthy subjects may help to identify biomarkers of early prediction for severe disease outcome.
The aim of this study was to determine the association between genetic risk factors of severe influenza infection related to SNPs in hypercytokinemia genes and the plasma proteome markers in Canadian healthy young adults.
Subjects were from the Toronto Nutrigenomics and Health Study and included Caucasian men and women (n = 433) who completed a general health and lifestyle questionnaire and provided a fasting blood sample. Polymorphisms in the TNF (rs1800629), CCL1 (rs2282691), IL8 (rs4073) and LTα (rs909253) genes were extracted from an Affymetrix 6.0 chip and 54 plasma proteins were assayed by a mass spectrometry-based multiple reaction monitoring method.
The frequency of risk genotypes of rs2282691 (AA), rs4073 (TA), rs909253 (AG) and rs1800629 (GA) were, 42%, 44%, 40% and 24%, respectively. Participants were separated into four risk subgroups (zero-, one-, two- and ≥ three-risk genotypes) based on the number of risk genotypes of the four examined SNPs. Eighteen proteomic markers were significantly correlated with the number of risk genotypes. Several of these proteins were involved in the innate immune pathway such as haptoglobin-β chain (r = 0.34, p = 0.039), complement factor B (r = 0.32, p = 0.05), complement C9 (r = 0.31, p = 0.049) and downstream acute phase reactants such as CRP (r = 0.33, p = 0.043) and hemopexin (r = 0.34, p = 0.039).
Polymorphisms in genes related to hypercytokinemia and risk of severe influenza infection are associated with a proteomics profile linked to biomarkers involved in the innate immune system. This outcome may permit identifying public health measures for early risk prediction and prevention of severe outcome upon infection with influenza virus.
Hypercytokinemia, Influenza, Genotype, Proteomics, Nutritional intervention
Influenza type A Virus (IAV) is one the most pathogenically communicable viruses affecting humans in which genetic reassortment of viral proteins allows for antigenic shifts that can cause serious pandemics when human to human transmission occurs [1]. Upper respiratory tract infections can be mild and often self-limiting but can spread aggressively with the ability to induce severe complications, morbidity and mortality in a vulnerable or "at-risk" subset of the population [2,3]. Yearly seasonal influenza outbreaks cause more than 200,000 hospitalizations and 41,000 deaths in the United States where it is the seventh leading cause of death [4]. In Canada, influenza accounts for the highest incidence of infection in children under 2-years of age and is the eighth leading cause of death [5]. Serious complications of IAV infections can impact the pulmonary, cardiac and neurological systems, which can have detrimental outcomes specifically in genetically predisposed subjects [1,3].
Recent studies have demonstrated that hypercytokinemia is the principal immunopathological mechanism that contributes to severe clinical presentation in patients with influenza infections. The genes that code for pro-inflammatory cytokines and chemokines responsible for hypercytokinemia include TNFα, IFNγ, IL-1, IL-6, IL-8, IL-9, IL-12, IL-15, and IL-17 [6]. These genes are polymorphic and certain alleles have been associated with susceptibility to a wide range of infectious diseases as well as their severe outcome [7-9]. Although these mediators are principally associated with immune reactions, they also influence functions in epithelial and endothelial cells, smooth muscle, and adipose tissue [10] and play a role in sepsis, shock, acute respiratory distress, and responses to toxic medication [10,11]. Clinical signs and symptoms associated with hypercytokinemia include headaches, muscle pain, nausea, diarrhea, vasodilatation, and hypotension [10] and can be responsible for the symptoms caused by IAV [12].
Several variants in the genes encoding these cytokines, e.g., TNF (rs1800750 G/A), IL1B (rs16944 G/A), CCL (rs2282691 A/T) and IFITM3 (rs12252 T/C) have been associated with risk of severe clinical manifestations in IAV [13]. Furthermore, elevated levels of the resultant pro-inflammatory cytokines and chemokines were found in the plasma of patients with acute respiratory distress syndrome caused by IAV [14,15]. Previous studies examining the relationship between infectious diseases and cytokine markers suggest that host genetic factors may indeed influence these biomarkers and the subsequent disease severity [16], as was also noted in severe IAV infection [14,15,17,18]. Advances in proteomics enable the simultaneous detection of multiple proteins in plasma that assist in understanding the interactions within and between pathways. Analysis of the genetic variants in cytokine genes related to severe IAV, when examined in association with the profile of the affected proteins, may identify novel proteins related to risk of disease severity. The aim of this study is to determine the association between variants in genes coding for cytokine synthesis and the downstream proteomic biomarkers in a young Canadian population to identify novel proteins related to genetic risk of severe influenza upon infection.
The Toronto Nutrigenomics and Health Study is an ethnoculturally diverse population of men (n = 520) and women (n = 1,117) at 20-29 years of age, recruited from the University of Toronto campus. All subjects completed a general health and lifestyle questionnaire, which collected information on subject characteristics including age, sex, medical history, smoking status and ethnocultural group. Information on habitual dietary intake over the past month was collected using a semi-quantitative food frequency questionnaire, modified from the Willett questionnaire and included questions on supplement use, as previously described [19]. Fasting venous blood samples for biomarker assessment and DNA isolation were drawn from participants following a 12-hour fast. Average blood pressure measurements were taken at rest, twice at one minute intervals, using the OMRON IntelliSense Blood Pressure Monitor (Model HEM-907XL; OMRON Healthcare, Vernon Hills, Illinois) [20]. The study was approved by the Research Ethics Board at the University of Toronto and was in compliance with the Declaration of Helsinki. All subjects provided written, informed consent.
Subjects provided a blood sample after an overnight fast. Blood samples were collected and analyzed for biomarkers of cardiometabolic disease at LifeLabs Laboratories (Toronto, ON, Canada). Serum ascorbic acid was measured using High Performance Liquid Chromatography (HPLC) as described elsewhere [19]. The Homeostasis Model of Insulin Resistance (HOMA-IR) and beta-cell function (HOMA-β) were calculated from measures of insulin (µU/mL) and glucose (mmol/L). HOMA-IR was calculated using the formula insulin × glucose/22.5 and HOMA-β was calculated using the formula 20 × insulin/(glucose-3.5). Aliquots of plasma from sodium heparin- and EDTA-treated blood samples were shipped from LifeLabs to the University of Toronto (Toronto, ON, Canada) and stored at -80 ℃. Proteomic analysis was conducted at the Genome British Columbia Proteomics Centre at the University of Victoria (Victoria, BC, Canada) as previously described [19].
Genotyping from the Caucasian subset of the study population (n = 433; 120 males and 313 females) was performed through Genome-Wide Association Genotyping (GWAS) using the Affymetrix 6.0 Human SNP Array. SNPs in hypercytokinemia-associated genes were extracted from the GWAS chip and consisted of tumor necrosis Factor (TNF, rs1800629), chemokine ligand-1 (CCL1, rs2282691), interleukin-8 (IL8, rs4073) and lymphotoxin-alpha (LTα, rs909253). After excluding subjects with missing or incomplete proteomics data from blood samples, analyses were performed on a total of 399 Caucasian subjects (105 males, 294 females).
Statistical analyses were carried out using Statistical Analysis Software (version 9.4, SAS Institute Inc., Cary, North Carolina). The distributions of continuous variables were examined prior to analysis and loge- or square root- transformed to improve normality where necessary. Means and measures of spread are reported in untransformed states. For all analyses, the α-error was set at 0.05 and reported p values are two sided. Differences in subject characteristics between males and females were assessed using t-test for continuous variables and χ2 tests for categorical variables. Analysis of Covariance (ANCOVA) was used to compare mean biochemical characteristics by genotype adjusted for age, sex, BMI and Hormonal Contraceptive (HC) use among women. The study population was divided into four subgroups consisting of subjects with no- (n = 68), one- (n = 141), two- (n = 118) and ≥ three- (n = 72) risk genotypes. These risk groups were assigned solely based on the number - not the specific type - of risk genotypes. Comparison between group means was carried out by one-way ANOVA test. Comparisons between the no-risk and ≥ three-risk genotypes groups were conducted by t-test adjusted for age, sex, BMI and HC use. Among these risk groups, Pearson's correlation coefficient (r) between the levels of biochemical markers and the number of risk genotypes for all study subjects was calculated adjusted for age, sex and BMI. Similar correlation analysis was conducted between the individual SNPs and the proteomic markers. Departure of genotype distributions from Hardy-Weinberg equilibrium was assessed using a χ2 test with 1 degree of freedom.
The present study determines the association between variants in genes linked to severe influenza infection and code for cytokine synthesis and proteomic biomarkers in a population of young Canadian adults. The objective is to identify novel proteins related to genetic risk of severe influenza infection. Subject characteristics are shown in Table 1. Serum levels of the biochemical markers were all within the normal clinical ranges. LDL, HDL, total cholesterol, triglycerides, insulin and C-Reactive Protein (CRP) were significantly (p < 0.05) higher in females than males. In general, females had higher levels of vitamins such as ascorbic acid, α- and β-carotene, α-tocopherol, and vitamin D compared to males, but slightly lower levels of δ- and γ-tocopherol.
Table 1: Characteristics of study participantsa. View Table 1
Genotype frequency for CCL1 (rs2282691), IL8 (rs4073), LTA (rs909253) and TNF (rs1800629) are shown in Figure 1. The frequency of risk genotypes of rs2282691 (AA), rs4073 (TA), rs909253 (AG) and rs1800629 (GA) were 42%, 44%, 40% and 24%, respectively. All SNPs were in Hardy-Weinberg equilibrium (p = 0.686, 0.992, 0.481, and 0.919 for rs2282691, rs4073, rs90925, and rs1800629, respectively). Analysis of the relationship between genotypes of the examined SNP and the biochemical measures in the studied population (Table 2) adjusted for age, sex, BMI and HC use among women-revealed several significant associations. For example, CCL1 rs2282691 genotypes were significantly associated with total cholesterol, lutein, β-carotene, α-tocopherol and cis-5-lycopene. IL8 (rs4073) genotypes were associated with HOMA-IR and insulin as well as trans- and cis-lycopenes. Only lutein was significantly associated with the different genotypes of LTA (rs909253) whereas TNF (rs1800629) genotypes were associated with HOMA-Beta and δ-tocopherol.
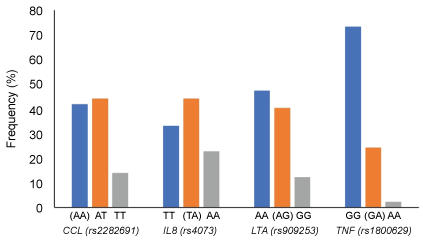 Figure 1: Genotype frequency for CCL1 (rs2282691), IL8 (rs4073), LTA (rs909253) and TNF (rs1800629) in the study population. Risk genotypes are shown in parentheses for each SNP [13]. View Figure 1
Figure 1: Genotype frequency for CCL1 (rs2282691), IL8 (rs4073), LTA (rs909253) and TNF (rs1800629) in the study population. Risk genotypes are shown in parentheses for each SNP [13]. View Figure 1
Table 2: Characteristics of the study subjects in relation to genotypes of hypercytokinemia genes. View Table 2
Based on the number - not the specific type - of risk genotypes, the study subjects were stratified into four risk subgroups (no-, one- two- and ≥ three-risk genotypes). One-way analysis of variance test showed a significant difference between these risk genotypes groups in the average levels of some nutritional factors such as δ-tocopherol and 25(OH)D, in inflammatory markers including CRP, ILK-1 receptor antagonist and PDGF-BB, the innate immunity complement pathway (complement C4 γ chain) and in protein markers such as afamin, apolipoproteins A-II precursor and B-100 and beta-2-glycoprotein (Table 3). We examined the difference between the no- and ≥ three-risk genotypes groups and the correlation between the protein markers and the risk subgroup (Table 3). There was no association between the individual SNPs and the various proteome markers (data not shown). However, the correlation between biochemical, nutritional and proteomics markers and risk subgroup indicated a significant association with 18 proteins, several of which are involved in the innate immune pathway. These included the cascade activator haptoglobin-β chain (r = 0.34, p = 0.039), members of the complement pathway such as complement factor B (r = 0.32, p = 0.05) and complement C9 (r = 0.31, p = 0.049), and downstream acute phase reactants such as CRP (r = 0.33, p = 0.043) and hemopexin (r = 0.34, p = 0.039) in addition to L-selectin (r = 0.39, p = 0.021), a mediator for leukocyte function. Significantly higher levels of δ-tocopherol were found in the ≥ 3 risk genotypes group compared to the no-risk group (p < 0.01). This difference was coupled by a significant correlation between the number of risk genotypes and levels of afamin (r = 0.36, p = 0.046) that plays a role in vitamin E transport [21].
Table 3: Levels of proteomic and inflammatory markers in relation to risk genotypes of hypercytokinemia genesa. View Table 3
Previous studies have examined the proteomic profile associated with the response to IAV infection and the heritability of risk of severe disease in Mexican, Chinese, African and American Indigenous populations [13, 22-25]. However, studies comparing the association between variants in cytokine-coding genes and various proteome biomarkers of risk for severe infection are lacking in the general healthy population and in Canadians. To our knowledge, this is the first study to examine the association between SNPs in hypercytokinemia genes and plasma proteomics markers in a healthy Canadian population.
Risk alleles and the specific genotypes identified in polymorphic genes of Mexican Mestizos during the 2009 flu pandemic include the AG genotype at rs1800750 in TNF, AG genotype at rs16944 in IL1B and the AA genotype at rs2282691 in CCL1 [13]. In addition, the AT genotype at rs4073 in IL8 [16] and the AG at rs909253 in LTα, also known as TNF-β [16,26], were associated with the inflammatory response [13]. In the present study, the genotypes identified to play a role in IAV, were also replicated to have similar effects in another study among a Greek population [27]. Such polymorphisms in hypercytokinemia genes are associated with an increased potential to synthesize cytokines and chemokines and influence the nature and effectivness of the inflammatory immune response [13,27]. Overall, risk alleles of IL8 and TNF-β were associated with a 1.5 to 3-fold increased risk of IAV infection and with a 1.2 to 2.4 increased risk of death [16,28]. This observation may facilitate the characterization of genetic factors and downstream protein biomarkers that can be employed in identifying individuals at high risk of disease severity upon and during infection. For example, in severe IAV cases, TNF rs361525 was more common in patients compared with controls [29]. Furthermore, diagnosis of pneumonia was 3-fold more common in cases with at least one copy of the minor allele than in cases with no copies [29]. Risk alleles of TNF rs361525 were found to primarily influence disease severity following infection rather than susceptibility to infection [29]. An ethnic-specific pattern of genetically pre-defined disease severity can be triggered by a range of environmental and nutritional factors and may result in phenotypic profiles related to disease severity [29].
We demonstrated an association between the number - not the specific type - of risk genotypes and protein factors involved in the innate immune pathway. These included the cascade activator haptoglobin β chain, complement factor B and complement C9, the downstream acute phase proteins such as CRP and hemopexin and L-selectin that mediates leukocyte function. There was also a significant difference between the risk genotypes groups in the average levels of inflammatory markers such as CRP, ILK-1 receptor antagonist and PDGF-BB as well as the complement C4 γ chain (Table 3). Attenuation of the innate immune system during infection can be genetically- and/or environmentally-mediated leading to the overproduction of pro-inflammatory cytokines such as ILs, TNF-α, IFN-γ and TGF-β [17,18]. This response is known to occur in a number of infectious diseases [30-32] including IAV [14,15,17,18], leading to the ensuing delayed clearance of viral load, vasculopathy, hemorrhage and tissue [14,15,17,18,33,34]. The results of the present study suggest that polymorphisms in the hypercytokinemia genes are associated with a proteomic profile linked to innate immunity activation (e.g., haptoglobin β chain), the modulation of the complement pathway, increased levels of acute phase reactants (e.g., CRP and hemopexin) and altered levels of L-selectin influencing the overall function of leukocytes. The cytokine overload related to the Th1 to Th2 shift in severe viral infection - when accompanied by increased cytokine synthesis arising from gene polymorphisms - both can be detrimental to the endothelium and lead to a range of subsequent complications [35]. The altered innate immune pathway and shift in Th1 (microbicidal action of IFN-γ) to Th2 (anti-inflammatory IL-4, -5, -10 and -13) may play a potential role in severe viral presentation as was noted by the high prevalence of, for example, allergy or eosinophilic responses observed in fatal viral infections [36]. It may be reasonable, therefore, to suggest that in infected patients, variants of the hypercytokinemia genes may synergistically attenuate the innate and humoral immune systems to further impair critical components of these pathways such as chemotaxis, phagocytosis, and the bactericidal activity of neutrophils and macrophages [30]. These outcomes may subsequently downregulate the functions of T cells and neutrophils to exacerbate the complications of the infectious diseases [30].
Significant increases in the levels of δ-tocopherol were found in the subjects with ≥ three-risk genotypes compared to those with "no-risk" genotype. This was particularly apparent in subjects with the risk genotype of TNF rs1800629 compared to their counterparts with other TNF genotypes. There was also a significant difference between the risk genotypes groups in the average levels of nutritional factors such as δ-tocopherol and 25(OH)D (Table 3). Moreover, γ-tocopherol, which is more abundant in the American diet than other forms of vitamin E (e.g., α-tocopherol), was non-significantly elevated in subjects with ≥ three-risk genotypes, but was significantly higher in the AA risk genotype of the CCL1 rs2282691. The observation that the number of risk genotypes is correlated with the level of afamin, a vitamin E-binding glycoprotein that plays a role in vitamin E transport [21], may shed some light on the potential relationship between the antioxidant characteristics of vitamin E and risk of severe clinical IAV [31,32,37]. Furthermore, consideration of vitamin E intake may also be beneficial for women who possess the cytokine risk genotypes given their overall lower levels of δ-tocopherol and γ-tocopherol compared with men. As with vitamin E, numerous studies have assessed the role of ascorbic acid intake during a common cold and have reported the reduction of lung inflammation caused by IAV when taken synergistically with other nutrients [38,39]. Large doses of vitamin C have been shown to relieve symptoms [40]. Increased intake of vitamin C and other antioxidants by the at-risk group, who also have lower ascorbic acid concentrations, may potentially have greater preventive benefits than in those with lower number of risk genotypes.
The present study has a number of limitations. Although diet and lifestyle factors affect protein levels and function, they were not considered in this study. Instead, we assessed the serum levels of various nutrients. The study also consisted of only Caucasians because of the small sample size of the other ethnic groups. Future studies are warranted to evaluate the association between the identified genetic risk factors of influenza and the associated phenotype in individuals diagnosed with severe infections. This will permit a better understanding for the utility of the risk profile characterized here and linked to an altered innate immune pathway in predicting severe disease outcome in asymptomatic subjects.
In conclusion, polymorphisms in hypercytokinemia genes are associated with an increased potential to synthesize cytokines and chemokines and influence the innate immune responses to play a role in severe influenza infection. This was noted, for example, in the effect on these genes on the levels on haptoglobin-β chain, complement factor B, complement C9 and the downstream acute phase reactants CRP and hemopexin. This observation was substantiated from the overall association between the risk genotypes and the proteomics profile related to the innate immune pathway. Identifying genetic and proteomic markers associated with risk of severe influenza infection may facilitate targeting these biomarkers as an effective approach for disease prevention, particularly in the "at-risk" subpopulation. Furthermore, elucidating the interrelationship between genetic and phenotypic risk markers may permit a better understanding of the potential preventive efficacy of micronutrients on the severe disease outcome. Such an approach may permit developing a range of public health measures and recommendations for early disease risk prediction and prevention prior to the onset of severe clinical presentation.
This work is supported by the Public Health Agency of Canada (AB).
AE holds the Canada Research Chair of Nutrigenomics. The authors would like to thank Joseph Jamnik for helping in data collection.
AB conceived of the study and contributed to data analysis and interpretation and drafting the manuscript. SBD preformed data analysis and interpretation and drafted the manuscript. AES supervised data collection, analysis and interpretation and drafting the manuscript. All authors participated in study design, data interpretation and provided critical manuscript revisions, read and approved the final manuscript.
The authors declare no conflicts of interest.
The study was approved by the Research Ethics Board at the University of Toronto and was in compliance with the Declaration of Helsinki.