The novel severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) has posed a serious threat to the global public health. Respiratory failure, followed by cardiovascular complications with wide-spread endothelial dysfunction and inflammation, is rapidly emerging as a key threat in COVID-19. ACE-2 receptors are the cell-entry gate for SARS-CoV-2. The purpose of the present study is to evaluate Valproic Acid (VPA) as a potential drug to treat COVID-19 and look into its mechanism of action.
We demonstrate that VPA-treatment significantly reduced ACE-2 expression in endothelial cells. VPA-treatment significantly reduced the expression of inflammatory cytokines IL-6 along with the endothelial activation marker ICAM-1.
We provide evidence and discuss the plausible mechanism in detail for VPA in its uses to prevent and treat COVID-19 in a personalized manner. Our study is expected to entice the scientific and clinical society to investigate VPA as a potential therapeutic option against COVID-19.
Valproic acid, Endothelial cells, EndMT, COVID-19, ACE-2
The pandemic of coronavirus disease 2019 (COVID-19) caused by the novel coronavirus severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) has posed a serious social and economic threat worldwide by infecting over 4 million and killing 276,000 people. The real course of the disease is still not well described. However, respiratory failure followed by cardiovascular complications with underlying inflammation and thrombus formation are rapidly emerging key threats in COVID-19 [1,2]. A "cytokine storm" or overproduction of pro-inflammatory cytokines such as interleukins (ILs) and tumor necrosis factors (TNFs) is reported in the lungs of COVID-19 patients [3]. Currently, there are no approved drugs or vaccines with proven clinical efficacy to treat or prevent COVID-19. However, the US Food and Drug Administration (FDA) approved limited emergency use for hydroxychloroquine (HCQ) [4,5]. Other drugs such as arbidol, remdesivir, and favipiravir are currently under clinical trial to treat COVID-19, with mixed reports on their efficacy in the treatment of COVID-19 [5]. There is an urgent need for an effective drug to treat and prevent COVID-19, with minimal side effects.
Limited knowledge about the mechanism of infection/action of SARS-CoV-2 appears to be the major problem in the identification of therapeutic targets and respective drugs to treat COVID-19. However, Gordon, et al. followed a comprehensive approach and cloned, tagged and expressed 26 of the 29 SARS-CoV-2 proteins and identified 332 high-confidence SARS-CoV-2-human protein-protein interactions [6]. The majority of SARS-CoV-2 interacting proteins were associated with replication, epigenetic regulation and vesicle trafficking pathways [6]. In their human lung mRNA expression profile, they identified enrichment of SARS-CoV-2-interacting protein and an epigenetic regulator, histone deacetylase 2 (HDAC2), which regulates epigenetics by removing acetyl groups from histones [6,7]. Many non-histone proteins such as transcription factors, chaperones and viral proteins are also subjected to acetylation [7]. Gordon, et al. also identified 66 therapeutic targets for 69 compounds that include valproic acid (VPA) [6]. VPA is a FDA-approved HDAC2 inhibitor drug used to treat central nervous system diseases such as bipolar disorder, epilepsy, and cancer [8-10]. VPA inhibits HDAC2 by inducing its proteasomal degradation [9].
There are unequivocal evidence that angiotensin-converting enzyme 2 (ACE-2) receptors are the 'entry door' for SARS-CoV-2 to infect cells [11,12]. ACE-2 receptors are mainly expressed on endothelial and epithelial cells [2,11]. Endothelial cells (ECs) contribute to more than 30% of all cells in the lungs [13], constitute the innermost layer of every blood vessel and respond to constantly varying hemodynamics to maintain homeostasis [14]. ECs are plastic in nature; they have the capability to lose endothelial characteristics and transition into 'stem cell-like" mesenchymal cells, a process known as endothelial-to-mesenchymal transition (EndMT) [15,16].
HCQ, which is being tested in various clinical trials against COVID-19, is an autophagy inhibitor [17]. Autophagy is a key homeostatic process, where cytosolic components are degraded and recycled through lysosomes for reuse [17]. We have previously demonstrated that inhibition of endothelial autophagy via genetic deletion of autophagy-related gene 7 (ATG7) or by pharmacologic inhibition with bafilomycin induces EndMT-like phenotypic switching in ECs [18]. Interestingly, a similar effect of VPA was also observed, where VPA induced EndMT-like phenotypic switching in ECs [19]. Given that ACE-2 is expressed basally and widely on the endothelial cells [2,11], we hypothesized that VPA-induced EndMT-like phenotypic switching causes reduced expression of ACE-2 and thereby would inhibit the SARS-CoV-2 rate of infection. We, for the first time, show that VPA downregulates ACE-2 expression and inhibits the expression of inflammatory cytokines in ECs. We also provide a detailed review on the mechanism of action of VPA and propose the plausible mechanism of the drug to protect and treat COVID-19 patients, encouraging personalized therapy.
Human umbilical vein ECs (HUVECs, pooled, Lonza) or human coronary artery EC (HCAECs, Lonza) were cultured in EC growth medium-2 (EGM™-2 Bulletkit™; Lonza) containing growth factors or MCDB 131 (Gibco) supplemented with serum and antibiotics. Following 60-70% confluence, the cells were starved over-night in 1% FBS and then treated with 5, 10 and 20 mM of VPA (Santa Cruz Biotechnology) for 24 hours as described [19-21]. The control groups were treated with the diluent. RNAs were extracted using Trizol (Invitrogen), and cDNA was synthesized (QuantiTect, Qiagen) according to manufacturer's instruction. Quantitative (q) PCR was performed to measure the expression level of genes using SYBR® Select Master Mix (Applied Biosystems) and primers for ACE-2 [20], IL-6 (Forward-5'-CATTGGAGCAAGTGTTGGATCTT-3' and Reverse-5'-GAGCTAATGCATGCCATTCTCA-3'), and inter-cellular adhesion molecule-1 (ICAM-1) [20] in QuantStudio™ 3 Real-Time PCR System. GAPDH was used as the internal control [20]. Fold gene expression was calculated using the 2-∆∆Ct method, and the expression level in the vehicle-treated cells was considered as 100% to calculate % expression in VPA-treated cells. The differences between the two groups were calculated using Student's T-test, and differences between more than two groups were calculated using one-way ANOVA with Tukey's test. A p-value of < 0.05 was considered significant.
To test our hypothesis, we evaluated ACE-2 expression in cultured HUVECs that were treated with clinically relevant dose (5 mM) of VPA [19,21] for 24 hours. HUVECs represent the standard cellular model to study endothelial cells in vitro [19,20,22]. Our qPCR data demonstrated a significant ~70% reduction (p = 0.0079) in the expression level of ACE-2 in VPA-treated ECs in comparison to control ECs (Figure 1A). Given the venous nature of HUVECs, we next evaluated the expression level of ACE-2 in arterial ECs by treating cultured HCAECs with 5 mM of VPA for 24 hours. Similar to HUVECs, we observed a significant ~70% reduction (p = 0.0159) in the ACE-2 expression in VPA-treated in comparison to vehicle-treated control HCAECs (Figure 1B). To assess the dose-response effect of higher doses of VPA on endothelial ACE-2 expression, we treated HUVECs with 10 mM and 20 mM of VPA for 24 hours and evaluated ACE-2 expression. Our qPCR data showed a significant reduction (p < 0.0001) in ACE-2 expression for both 10 mM and 20 mM of VPA, but there was no significant difference (p = 0.7485) in ACE-2 expression between the two doses in VPA treated ECs (Figure 1C). Next, we evaluated the effect of VPA on the expression of pro-inflammatory IL-6 and the endothelial activation marker ICAM-1. We observed a significant downregulation of IL-6 (p < 0.0001) following 5 mM of VPA treatment to HUVECs in comparison to control (Figure 1D). Interestingly, VPA also significantly reduced ICAM-1 expression (p = 0.0022) in VPA-treated ECs in comparison to control ECs (Figure 1E).
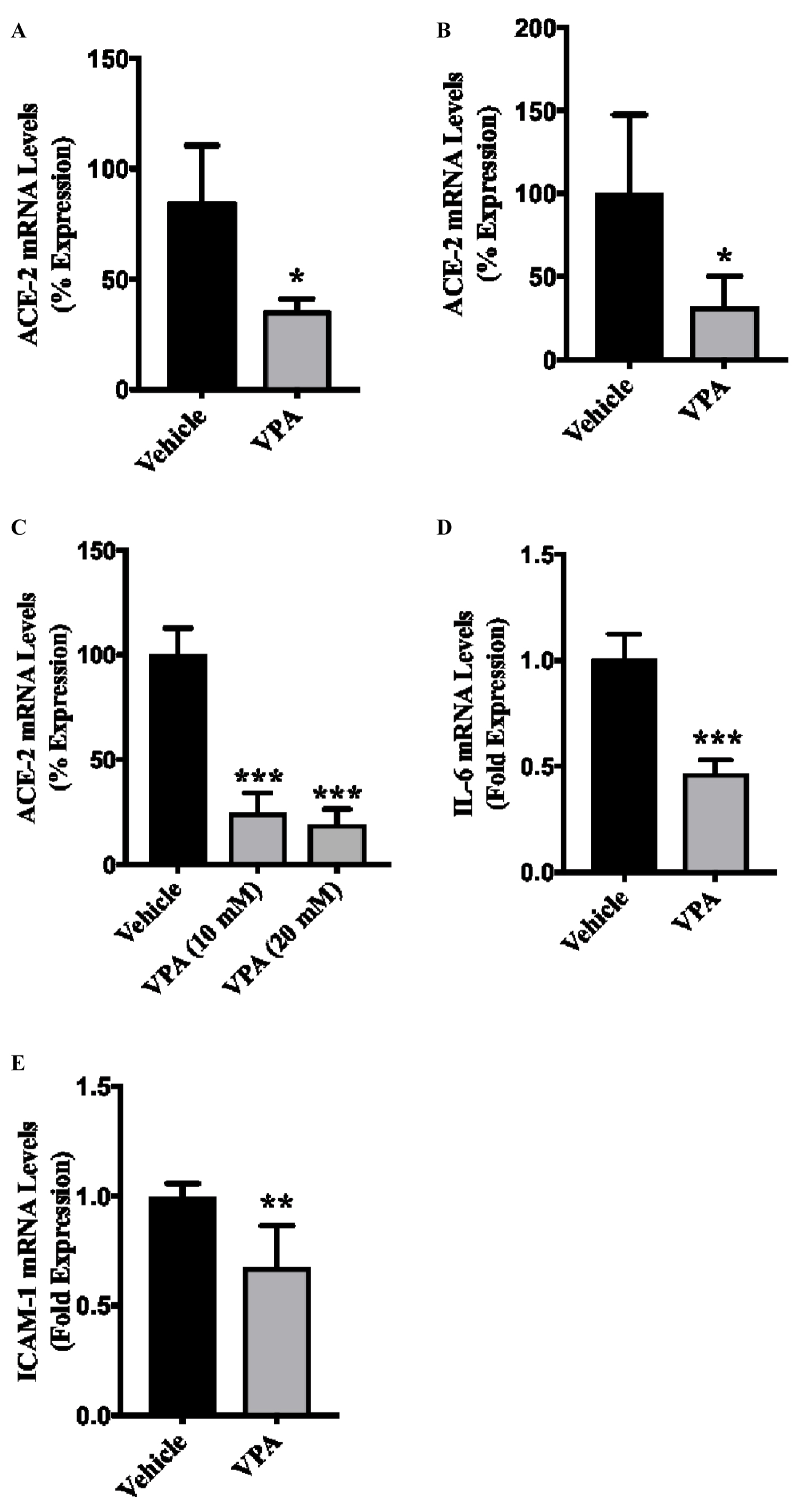 Figure 1: VPA-treatment downregulates ACE-2 and inflammatory molecule expression in ECs. HUVECs or HCAECs were treated with either diluent, or 5, 10 or 20 mM of VPA and total RNAs were extracted 24-hours post-treatment. (A, C) qPCR data demonstrating reduced ACE-2 expression in VPA-treated, HUVECs and (B) HCAECs; (D) VPA-treatment (5 mM) also reduced the expression of IL-6; and (E) ICAM-1 in HUVECs. N = 3-4 in triplicates.
Figure 1: VPA-treatment downregulates ACE-2 and inflammatory molecule expression in ECs. HUVECs or HCAECs were treated with either diluent, or 5, 10 or 20 mM of VPA and total RNAs were extracted 24-hours post-treatment. (A, C) qPCR data demonstrating reduced ACE-2 expression in VPA-treated, HUVECs and (B) HCAECs; (D) VPA-treatment (5 mM) also reduced the expression of IL-6; and (E) ICAM-1 in HUVECs. N = 3-4 in triplicates.
*p < 0.05, ** p < 0.01, ** p < 0.001 vs. corresponding diluent control group.
View Figure 1
VPA is a well-known HDAC2 inhibitor, which has been safely used for over 50 years as an anticonvulsant drug [8-10]. VPA is taken-up by endothelium immediately within a minute of intravenous injection in humans [23]. Cardiovascular complications with wide-spread endothelial dysfunction, thrombosis and endotheliitis are rapidly emerging as a key threat in COVID-19 in addition to respiratory disease [1,2]. The endothelium plays a central role in inflammation, thrombosis and cardiovascular complications. SARS-CoV-2 use ACE-2 receptors for cell entry, which are mainly expressed on ECs [2,11,12]. Altogether, these informations indicate a bigger role for VPA and endothelium in COVID-19. Accordingly, to devise therapeutic strategies to counteract SARS-CoV-2 infection and the associated pathology, it is crucial to understand and to apply the present published knowledge towards repurposing already approved drugs to treat or prevent COVID-19. Accordingly, we plan to evaluate the potential role of the drug VPA on ACE-2 expression in ECs. We also discuss our findings in relation to other published articles [19], providing a plausible mechanism for the use of VPA for the prevention and treatment of COVID-19 (Figure 2).
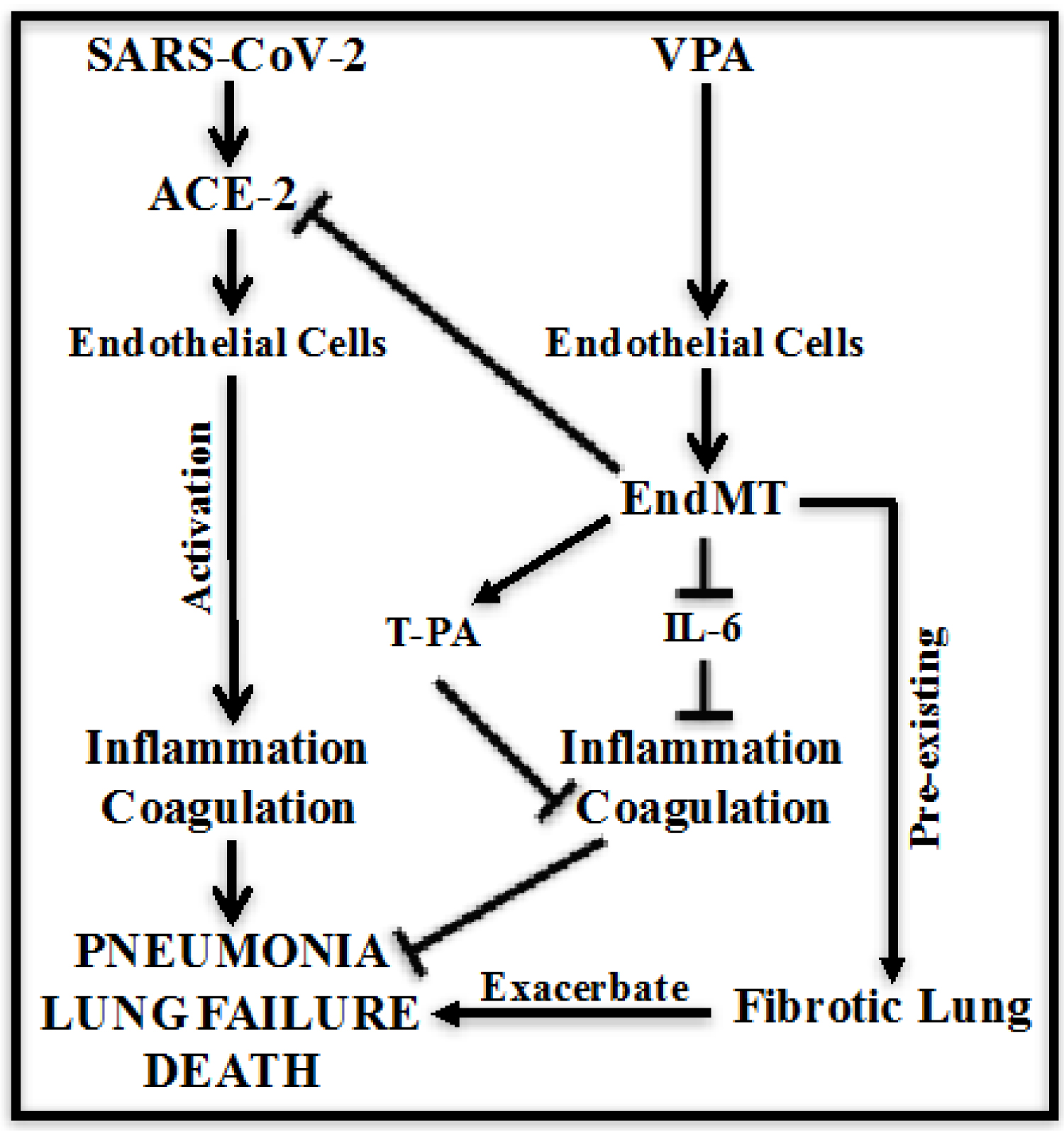 Figure 2: Schematic representing VPA-associated mechanisms in ECs to prevent and protect against COVID-19. VPA-treatment reduces ACE-2 expression, which is a gate-entry receptor of SARS-CoV-2 on EC. VPA also reduces inflammatory IL-6 expression along with endothelial activation marker ICAM-1, which will reduce thrombosis. VPA-induced T-PA expression will further protect against thrombosis, overall reducing SARS-CoV-2 rate of infection, SARS-CoV-2-induced inflammation and thrombosis to reduce pneumonia, lung failure and deaths in COVID-19 patients.
View Figure 2
Figure 2: Schematic representing VPA-associated mechanisms in ECs to prevent and protect against COVID-19. VPA-treatment reduces ACE-2 expression, which is a gate-entry receptor of SARS-CoV-2 on EC. VPA also reduces inflammatory IL-6 expression along with endothelial activation marker ICAM-1, which will reduce thrombosis. VPA-induced T-PA expression will further protect against thrombosis, overall reducing SARS-CoV-2 rate of infection, SARS-CoV-2-induced inflammation and thrombosis to reduce pneumonia, lung failure and deaths in COVID-19 patients.
View Figure 2
Our data demonstrate that VPA treatment to ECs significantly reduced ACE-2 expression. This data is clinically very relevant for COVID-19 as ACE-2 is a cell "entry door" for SARS-Co-2 and also because SARS-CoV-2 infection is shown to be enhanced by over-expression [24] and diminished by inhibition of ACE-2 [25-27]. The effect of VPA on ACE-2 expression indicates that VPA can inhibit the SARS-CoV-2 rate of infection by reducing its receptor ACE-2 expression level and can be used as a prevention strategy against COVID-19. Next, our data on the reduced expression of IL-6 in VPA-treated ECs is also clinically relevant, as the cause of death is the inflammation and thrombosis due to the "cytokine storm" mainly of interleukins such as IL-6 in the lungs of COVID-19 patients [3]. Particularly, the level of IL-6 predicts respiratory failure, and IL-6 inhibitors are proposed to ameliorate severe lung damage in COVID-19 patients [28]. ECs can secrete pro-inflammatory cytokines, and these cytokines "activate" endothelial cells to produce tissue factor, which regulates thrombosis [29,30]. ICAM-1 is markers for endothelial "activation" [30]. Interestingly, VPA also significantly reduced ICAM-1 expression, indicating reduced endothelial activation (Figure 1E). The presented data is of immense importance as it provides a "treatment" strategy for COVID-19 as VPA-induced downregulation of IL-6 and ICAM-1 are the regulatory molecules implicated in SARS-CoV-2-induced inflammation and coagulation.
PCR array analysis for human endothelial-related genes in VPA-treated ECs is reported with a total of 14 significantly up- and 14 down-regulated genes [19]. To evaluate the therapeutic role of VPA in COVID-19, we investigated these VPA-induced differentially expressed endothelial genes in relation to inflammation, coagulation, endothelial function, endotheliitis, cardiovascular and respiratory diseases. The most upregulated endothelial genes which are relevant to COVID-19 prevention and/or treatment include BNP (Natriuretic peptide B; 145-fold), MMP-9 (Matrix metallopeptidase 9, 121-fold), PF-4 (Platelet factor 4, 25-fold), T-PA (Plasminogen activator, tissue, 18-fold), COX-2 (Prostaglandin-endoperoxide synthase 2, 3-fold) and TGFβ1 (Transforming growth factor, beta 1, 1.3-fold). BNP was the most upregulated gene in VPA-treated ECs, which is a diagnostic biomarker for cardiac dysfunction [31], but is also anti-inflammatory and inhibit interleukins [32,33]. The next upregulated gene MMP-9 is anti-inflammatory [34,35] and is associated with lung remodeling [36]. PF-4 is anti-viral as it inhibits human immunodeficiency virus type 1 (HIV-1) infection [37], and also protects the lungs against bacterial infection [38]. VPA-induced endothelial and anti-thrombotic T-PA is of particular relevance to the COVID-19 patients. T-PA treatment is suggested for COVID-19 associated acute respiratory distress syndrome (ARDS) [39]. Furthermore, VPA is known to have protective effects in severe hemorrhage and ischemia-reperfusion injury [40]. Coagulopathy has become a hallmark of severe COVID-19 with high rates of central line thrombosis and vascular occlusive events (e.g., ischemic limbs, strokes, etc.) [39]. Ventilator-induced lung injury is another cause in COVID-19 death, and T-PA is known to attenuate ventilator-induced lung injury [41]. Overall, VPA-induced T-PA appears to be beneficial to COVID-19 patients due to its anti-thrombotic activity and attenuation of ventilator-induced lung injury function. COX-2 was also upregulated in VPA-treated ECs. Given the important role played by COX-2 in antipyretic nonsteroidal anti-inflammatory drugs (NSAIDs), caution should be taken while prescribing NSAIDs with VPA to COVID-19 patients [42]. The most downregulated and relevant to COVID-19 prevention and/or treatment genes include TFPI (Tissue factor pathway inhibitor, 6-fold) and SPHK (Sphingosine kinase 1, 5-fold). Tissue factor is an initiator of coagulation and inflammation in the lung [43]. Most downregulated anti-coagulant TFPI activities are reported in the lungs of idiopathic pulmonary fibrosis patients [44]; they are also a marker for the prediction of deep venous thrombosis and tumor metastasis in patients with lung cancer [45]. Given the increased rate of thrombosis in COVID-19, the status of TFPI needs to be investigated with VPA-treatment. The VPA-induced downregulation of SPHK1 appears to help COVID-19 treatment as followed: SPHK1 is known to contribute to ventilator-associated lung injury [46]; elevated SPHK1 enhances influenza virus infection [47]; SPHK1 also serves as a pro-viral factor by regulating viral RNA synthesis and nuclear export of viral ribonucleoprotein complex upon influenza virus infection [48]. VPA-induced downregulation of SPHK appears to be helpful in the treatment of COVID-19 patients.
The exact mechanism behind HCQ-mediated therapeutic benefit to COVID-19 patients is not completely elucidated, but what we know as a fact is that HCQ is an autophagy inhibitor [17]. TGFβ-signalling induces EndMT [16], and loss of endothelial autophagy [18] or VPA-treatment to ECs [19], both exhibit TGFβ-induced EndMT. It is quite possible that EndMT-associated loss of endothelial function and gain of pro-fibrogenic mesenchymal function is the mechanism behind HCQ-associated benefit in COVID-19 patients. In that scenario, similar benefits can be expected from VPA in COVID-19 patients. EndMT is known to play roles in the development process [49], and also in diseases such as pulmonary vein stenosis, anomalous vascular remodeling, cerebral cavernous malformations, cancer progression and organ fibrosis [50,51]. Most importantly, it was reported that 16% of lung fibroblasts in the pulmonary fibrosis mouse model originated from ECs through the EndMT process [52], and loss of endothelial autophagy-induced EndMT exacerbated lung fibrosis in the same mouse model [18]. The role of EndMT in exacerbation of lung fibrosis might be the reason behind the no-effect or adverse effect of HCQ in critically-ill COVID-19 patients; however this needs to be evaluated. Similar side effect can be expected for VPA, and if true, precautions should be considered before prescribing VPA to COVID-19 patients with pre-existing pathologies. Furthermore, VPA is an anti-seizure drug, and there are reports suggesting that patients with pre-existing seizure disorder may be at greater risk for getting breakthrough seizures [53]. In these COVID-19 patients, VPA can be a preferred treatment option.
This new knowledge about the mechanism of action of VPA provides a novel potential therapeutic drug target for the prevention and treatment of COVID-19, and warrants immediate further investigation in animal models and in humans. We also suggest that VPA based therapeutics may exacerbate EndMT-related pathologies in COVID-19 patients with pre-existing diseases such as lung cancer or lung fibrosis, and promote personalized therapy in such COVID-19 patients. Overall, our manuscript provides a better source of necessary knowledge on VPA for researchers and health care practitioners to find treatment for COVID-19. VPA is a FDA approved drug and any proof of these drugs showing protective effect against COVID-19 will reduce the required drug approval time. In addition, VPA can be fast-tracked for testing against COVID-19.
This work was supported by a grant from the Heart and Stroke Foundation of Canada (G-17-0018688) to KS. KS is also the recipient of the 2018/19 National New Investigator Award- Salary Support from the Heart and Stroke Foundation of Canada, Canada.
KS conceived and designed the study. SS helped with data analysis, figures, discussion and manuscript writing.
None.