Amyloidosis is characterized by extra-cellular deposition of an insoluble fibrillary protein, amyloid in organs and tissues. It is classified as either primary or secondary based on the presence of concomitant diseases, ranging from chronic infection or inflammation to malignancy and as limited or systemic based on the extent of organ invasion. The clinical and radiological manifestations of amyloidosis are varrious and often nonspecific, making amyloidosis a diagnostic challenge. Here, we present an asymptomatic local primary pulmonary amyloidosis case, diagnosed during an operation preparation.
Amyloidosis, Lung, Primary
Amyloidosis is a disease characterized by the accumulation of a non-soluble substance in the extracellular matrix of the organs and tissues. This substance is in the fibrillary protein structure [1-2]. It can be primary that means no related disease present or secondary hat is related with chronic infection, inflammation or malignancy [1-5]. Pulmonary amyloidosis can be a part of a systemic amyloidosis or it can be limited to only lung. Diagnosis is difficult because the clinical and radiological features are variable and usually nonspecific. We present an asymptomatic case who is diagnosed incidentally during a cholecystectomy operation preparation.
A 55-year-old male patient; was sent to the chest diseases clinic because of abnormal chest X-ray findings during preparation for cholecystectomy operation. The patient had no active respiratory complaints. He had no other special feature other than 50 pack/year smoking in his medical history. Blood pressure: 130/75 mmHg, pulse rate: 73/min and rhythmical, respiratory rate: 15/min, SpO2: 97% and no pathological findings in the respiratory system. He had bilateral radiopacity which was more pronounced in the upper zones in chest X-ray. In thorax computed tomography (CT), there were peripherally located consolidations and occasional glass ground appearances. These abnormalities were more prominent in the upper lobes rather than lower lobes. Respiratory function test results; forced vital capacity (FVC): 2.63 L (70.8%), forced expiratory volume in 1 second (FEV1) 2.19 L (71.3%), FEV1/FVC 82.68, and diffusing capacity of the lungs for carbon monoxide (DLCO) 75%.
Routine biochemical tests, hemogram and complete urinalysis showed no abnormalities. (White blood cell 13.10, Red blood cell 5.22, Hematocrit 42%, Hemoglobin 15.4, Sedimentation 16 mm/s BUN 12, creatinine 1.02 mg/dL, total protein 33 mg/day). Bronchoalveolar lavage (BAL) from the right upper lobe and transbronchial biopsy were obtained. In the BAL fluid, 10% macrophages, 10% lymphocytes and 80% neutrophils were observed. Transbronchial biopsy was reported to be compatible with amyloidosis with features such as amyloid-positive staining (crystal violet), interstitial edema, chronic lymphocytic infiltration and anthracosis. For the other system scan, the patient was consulted with the internal medicine department. Qualitative and quantitative serum immuno-electrophoresis analysis and k/λ were normal. (IgA: 351 mg/dL, IgM: 535 mg/dL, IgG: 1420 mg/dL, free kappa light chain in urine: 0.77 mg/dL, free lambda light chain in urine: 0.07 mg/dL). Bone marrow biopsy of the patient was reported as normocellular bone marrow with megakaryocyte hyperplasia. Ultrasonography of abdomen was normal. Transthoracic echocardiography was normal also. Patient was diagnosed as primary lung limited pulmonary amyloidosis and followed-up without treatment Figure 1 and Figure 2.
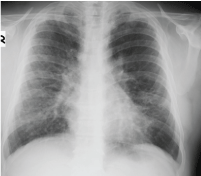 Figure 1: Image from chest X-ray.
View Figure 1
Figure 1: Image from chest X-ray.
View Figure 1
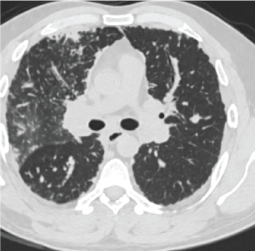 Figure 2: Image from CT scan.
View Figure 2
Figure 2: Image from CT scan.
View Figure 2
Amyloidosis is a rare disease that occurs due to the deposition of abnormal fibrillary protein, amyloid, that is supposed to result from incorrect protein folding, in any organ or tissue. It may be fatal or be detected incidentally with no complain [1-3,6].
A pulmonology can come across with a pulmonary amyloidosis patient either as part of systemic amyloidosis or as local amyloidosis, limited to only the respiratory tract. Pulmonary amyloidosis can be either primary, that means no other related disease is present or secondary that means it is related with another disease. In the United States, 1% of patients with chronic inflammatory conditions (rheumatic disease, infections, familial Mediterranean fever, malignancies) will develop secondary amyloidosis. According to the National Center for Health Statistics, the incidence of primary amyloidosis is 4.5 per 100,000 [6]. Amyloid can accumulate in the trachea, bronchia, parenchyma of the lung, or mediastinal lymph nodes. Parenchymal amyloidosis is the most common clinical condition among the amyloidosis that involve the respiratory tract. Parenchymal amyloidosis can be in nodular form or diffuse alveolar form [1-7]. Nodular form is usually seen in the secondary amyloidosis and may also be seen in primary amyloidosis as it is in our case.
Chest X-ray is often normal in amyloidosis. Bilateral, peripheral, subpleural nature, lower zone dominance and sizes between 0.5 cm and 15 cm in diameter are the general features of amyloidosis-related parenchymal nodules [1,3]. These features are valid for our case too except upper zone dominance. It is often diagnosed incidentally. It is important to diagnose these parenchymal nodules, as these can be related with an infection, a neoplasm or vasculitis [1-5]. Metastatic pulmonary calcifications can also be seen due to chronic renal failure if it is secondary to renal failure [2]. Calcifications due to renal failure are mostly seen in the upper lobes and central.
In the pulmonary function test, a restrictive pattern is expected mostly, but not always [1,7]. Comprehensive pulmonary function tests were performed in our patient. Light restriction and DLCO reduction were detected.
For the diagnosis, amyloid deposition must be histologically demonstrated. It has double break with congo red in green color polarized light microscopy [1,3,6,7]. In the pathologic examination of the transbronchial biopsy specimen, it was reported as a tissue compatible with amyloids positively stained with crystal violet stain.
We did not find any cause for systemic amyloidosis in our case. Multiple myeloma, waldenström macroglobulinemia and monoclonal gammopathy were excluded with the aid of protein electrophoresis. Hematological, biochemical and urine excretions were performed for systemic diseases. Bone marrow analysis was reported as normal. Present findings revealed primary pulmonary amyloidosis, limited only to the lung.
Pulmonary amyloidosis has a variable clinical course. The nodular form usually progresses slowly and rarely requires intervention. In Cordier, et al. study, 10% mortality was detected in patients with primary pulmonary amyloidosis [7]. Unfortunately, there is no accepted treatment for the disease. Clinical trials are inadequate to guide the treatment of symptomatic nodular amyloidosis. Treatment of systemic amyloidosis is chemotherapy directed against the underlying B-cell clone. Melphalan, prednisone, and colchicine may prolong survival in primary amyloidosis and there are some authors that advise to follow-up in parenchymal limited cases [2,7]. Since our patient was asymptomatic, we decided to follow up the clinical status and physiological parameters, without any treatment.
As a result, amyloidosis due to defective protein folding may be fatal or diagnosed as incidentally. As the most important disease is malignancy in the differential diagnosis, all patients are recommended to take histopathological diagnosis.