It remains a wonder how Tregs induce tolerance for the development of cancer. Previously we have shown with melanoma patients that, increase in peripherally induced Tregs (pTregs) number in blood is related to the poor prognosis of the disease. In vitro induced Tregs (iTregs) and pTregs are remarkably similar and significantly different in functionality from tTregs. Here, we worked with 12 melanoma patients- six HLA A2 positive and six HLA A2 negative. PBL and tumor cells were obtained from the patients with informed consent. Treg cells were generated and isolated from four different culture conditions: 1) Isolated from tumor +PBL IVC, 2) Mart-1 A2 or 3) Flu A2 pulsed DC + PBL IVC and 4) purified CD4+CD25- cells stimulated with anti CD3 and antiCD28 plus IL-2. We used these different Treg generation conditions (self vs. non-self) to understand how induced Tregs behave phenotypically and functionally that would open number of avenues to over come their negative effects. Here we show some phenotypic and functional characteristics of the induced Treg (iTreg) cell in cultures with PBL from the patients. We analyzed those Tregs for their suppressive function in separate CTL generation assays. We observed that iTreg cells under different conditions do not uniformly express CD25, FoxP3, PDL-1 or CTLA 4 as the known surface markers. When analyzed for their functionality, with adjusted number of cells, in suppressing the anti tumor CTL response, a significant difference was observed. The most effective Tregs cells were found to be those isolated from autologous tumor +PBL IVC or from Mart-1 peptide pulsed DC + PBL IVC. Those cells completely blocked the CTL induction and secreted huge amount of IL-10 upon re-stimulation. Further analysis with these different types of iTreg cells in terms of various gene expressions and corresponding protein secretion will be useful to find a target molecule to block such expansions of iTregs or pTregs cells for better therapeutic outcome.
Treg: T Regulatory Cells; tTre: Thymus Derived Natural T Regulatory Cells; pTreg: Peripherally Induced T Regulatory Cells; iTre: In Vitro Induced T Regulatory Cells; PBL: Peripheral Blood Lymphocytes; DC: Dendritic Cells; IVC: In Vitro Co Culture; TAA: Tumor Associated Antigen
It has been established that immune system generates specific cytotoxic T lymphocytes (CTL) to fight cancerous growth. Tumor associated (cancer) antigens are self-antigens and since the possibility for self-destruction by these Tumor antigen specific CTL is high, several regulatory activities exist to prevent autoimmune destruction by these CTLs [1-3].
Among all the known mechanisms of immune evasion by tumor cells, tumor associated antigen-specific T-cell tolerance through pathways of self-tolerance generation represents the significant challenge to successful immunotherapy for cancer [1,2]. CD4+CD25+ FoxP3+ T cells, referred to as T regulatory (Treg) cells, are selected in the thymus as a controller of anti-self repertoire [1,4-8]. These Tregs are referred to as natural T regulatory (nTreg) now called tTreg cells, are believed to down regulate anti tumor CTL response [2]. There is another class of CD4+ Treg cells also involved in regulatory function in the periphery, also phenotypically CD4+CD25+, and classified as induced Treg (iTreg), now called pTreg) cells [2,3]. These two different Tregs differ in their requirements for activation and in mode of action, but both are involved in the control of anti tumor CTL responses. The challenges are now to determine the effectiveness and the specificity of these Treg cells in recognizing the same tumor associated antigen (TAA) as that of the CTL population and to mitigate their regulatory constraints in order to maintain a robust CTL response against growing tumor [1,4-6].
In our previous reports we have shown that when CTL precursors with specificity for a self but tumor-associated epitope are optimally stimulated, tTreg are not much of an impediment but iTreg or pTreg are [9-12]. Our previous results also indicated that the freshly isolated Treg cells consist of a heterogeneous group of CD4+ T cells (both tTreg and pTreg) [13]. In order to block the negative role of pTreg cells on anti-tumor immunity there is a need for identifying the epitopes recognized by the pTreg cells in the tumor-micro-environment for their own expansion. In the present work we show suppressive function of TAA (self antigen) specific induced Tregs are antigen specific in specific CD8+CTL generation against TAA.
Melanoma patients were included in the study with appropriate approval from the institute's regulatory board. Blood samples from HLA A2 positive donors were taken for the study with informed consent. Patients diagnosed with primary cutaneous melanoma with or without systemic metastasis were included in the study. Informed consent was obtained from patients to donate 50 milliliters peripheral blood. If available and permitted by the pathologists, surgically removed tumor tissues were also obtained. The details of the collection of samples and cell preparation have been described [9,10].
Briefly, tissue cultures were performed in Iscove's medium (Life Technologies, Inc., Grand Island, NY) supplemented with 10% fetal bovine serum (FBS) (Gemini Bio-products, Inc., Calabasas, CA), L-arginine (0.55 mM), L-asparagine (0.24 mM) and L-glutamine (1.5 nM), (Life Technologies, Inc., Grand Island, NY) henceforth described as complete medium (CM). All the tissue culture plates were kept at 37 degree in CO2 (5%) incubators.
CD4+, CD8+, CD4+CD25+, CD4+CD25- T cells were purified using Magnetic beads as per protocol (Dynal Biotech, ASA, Oslo, Norway) and described earlier [8]. Briefly PBL were washed with cold PBS (4 ℃) with 2% FBS. Respective antibody labeled magnetic beads were add to the cell suspensions with cells: beads = 1:10. Incubated in ice for 30 min. Then we separated cells attached to the beads, by a magnet. A soln. of detach a bead were added to the mixture to free the positively selected cells from the beads.
Dendritic cells (DC) used in this study were isolated from peripheral myeloid antigen presenting cells (APC) expanded in vitro as described earlier [8,9,11]. Briefly, monocyte/macrophages were isolated as adherent cells from Ficoll Hypaque gradient derived mononuclear cell populations from blood. The adherent cells were then cultured in CM containing 1000 U/ml of GM-CSF (Immunex Corp., Seattle, WA) 1000 U/ml of IL-4 (R & D System, Minneapolis, MN) for 7 days. The non-adherent and loosely adherent cells were harvested by vigorous washing.
PBL or CD8+ T cells were co cultured with antigen pulsed DC or with autologous tumors as described earlier in several reports from our laboratory [10-13]. Briefly, tissue cultures were performed in iscove's medium (CM). Fresh tumor cells were isolated by mechanical fine mincing of tumor tissues in serum-free medium. Cells, stimulators and responders (1:10), were mixed in a 24 well or 12 well plates in CM with 50-100 u/ml of IL-2 and incubated 37 ℃ with 5% CO2 for specific number of days as needed for the experiments.
The immunofluorescence procedure for phenotypic analysis by flowcytometry has been described [8]. Briefly, for MHC class I HLA-A2 specific antibody from One Lambda Inc. (Canoga Park, CA) and for MHC class II antibody from BD Biosciences (San Jose, CA) were used, and the respective expression(s) on tumor cells and DC were studied by acquiring the cells using fluorescence-activated cell sorter machine (FACS): Facs Calibur (Becton Dickinson, Mountain View, CA) and the data were expressed as histogram overlay representing mean fluorescence intensity, using Cell quest and flowjo software (Becton Dickinson).
Cytokine, IFNg, IL-4 and IL-10 were assayed in appropriate ELISA assays as per manufacturer's (DuoSet ELISA Development System, R&D System, Minneapolis, MN) protocol and described earlier, Intracellular cytokine assay was done as described in our previous report [9-11]. Briefly, the effector cells were stimulated with peptide-pulsed DC for a total period of 6 hr at 37 ℃ (responder to stimulator ratio = 10:1). Two hours after initiation of the culture nonstimulatory doses of PMA (0.5 ng/ml) and Ionomycin (4 ng/ml, Sigma, St Louis, MO) were added for an additional 4 hr at 37 ℃. Brefeldin A (1 lg/ml, Sigma) was added 2 hr before harvesting the cells. Cells were washed and stained with FITC or PE-conjugated anti-CD8 antibody (Pharmigen, San Diego, CA) for 30 min on ice. Cells were then fixed for 25 min with 4% paraformaldehyde (Sigma) and permeabilized with PBS/BSA/Saponin for 10 min. Cells were then stained with human anti-IFN antibody conjugated with Allophycocyanin (APC; Pharmingen, San Diego, CA), and analyzed on FACS using Cell Quest or flowjo Software. Data were analyzed after acquiring 10,000 viable cells in each point.
Analysis for the MART-127-35 antigen specific and Influenza MP58-66 antigen specific TCR bearing CD8+ cells has been described previously [11]. Briefly, the cells from in vitro co cultures (IVC) were washed twice in phosphate-buffered saline (PBS) and then labeled with Mart-1 or Flu Allophycocyanin conjugate HLA-A2 tetramer (Beckman Coulter, Inc., Fullerton, CA) and CD8 FITC or PE (BD Biosciences) at room temperature for 30 min. The cells were again washed twice, re suspended in FACS buffer. Thereafter, the number of tetramer-positive cells was determined using FACS.
Cell mediated cytotoxicity assay was done following the method as described in our previous report [11]. Briefly, the 51Cr release micro cytotoxicity assays were done with freshly prepared or cryopreserved target cells were labeled with 51Cr with good efficiency. Target cells T2 or tumor cells were labeled with 51Cr for 30 min at 37 ℃. Target cells were washed with cold PBS three times and re suspended in medium and mixed with effector cells at different effector: target ratios in a 96 well V bottom plate with 200 ul in each well. Plates were incubated at 37 ℃ for four hours. Supernatant was collected and radioactivity released was counted in an appropriate radioactivity counter. The spontaneous release of the radioactivity from the 51Cr-labeled targets was usually < 12%. All the experiments in cytotoxicity assay were carried out in triplicate in the presence of 50-fold excess of cold K-562 cells to overcome the nonspecific killing activity.
We generated autologous tumor specific CTL with non HLA A2 patients' PBL (6/6 patients, UC1 to UC6) by stimulating PBL with autologous tumor. We also generated CTL with HLA A2 positive patients (UC-7 to UC-12) with Mart-1 (AAGIGILTV) or Flu peptide (GILGFVFTL) pulsed autologous DC. Table 1 shows the induction of CD8+ CTL from PBL IVC with nonA2 patients in cytotoxicity assays using autologous tumor cells as target cells. Cytotoxic effects were from the expanding CD8+ cells as the effect was blocked in the presence of anti CD8 antibody (data not shown here). Induced cytotoxic effector cells were very specific against autologous tumors cells. Figure 1a shows an example of tumor specific Mart-1 CTL induction form the cultures with peptide pulsed DC with HLA A2 patients' PBL. Figure 1b is an example of Mart-1 and Flu specific CTL generation with purified CD8+ cells. Mart-1 or Flu tetramer assays were used for determining the expansion of Mart-1 and Flu epitope specific CD8+ T cells. Although there were variations in the precursor frequencies for the two epitopes Flu from 0.05-1.4 and Mart-1 from 0.01 to 0.50 (data not shown) amongst individual donors, but when compared for fold expansion of the respective epitope specific precursors following stimulation, both Flu and Mart-1 epitope specific precursors expanded remarkably well after primary stimulation (Figure 1b). Figure 1c shows, specific precursors for autologous tumor cells and both the epitopes exhibited substantial expansion following antigen stimulation. Figure1d shows antigen specific secretion of IFN-γ by the different CTLs induced in IVC. It appears that antigen specific CD8+ CTL could be generated in culture using total PBL or purified CD8+ cells.
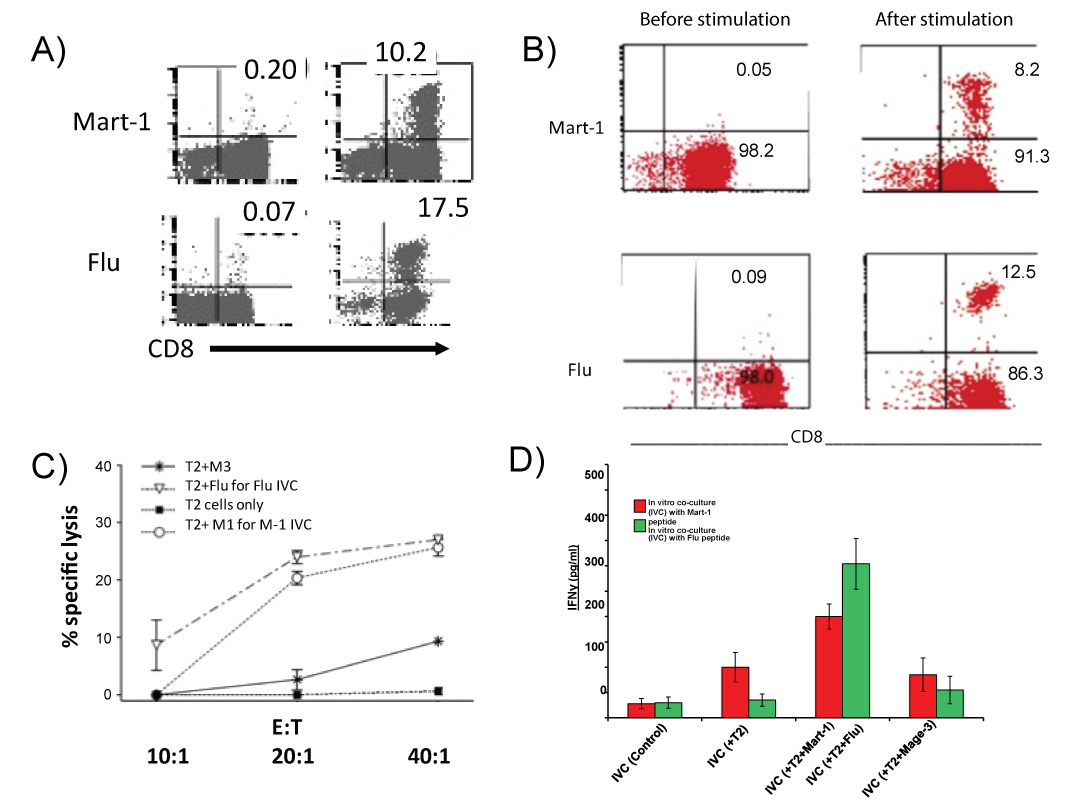 Figure 1: Expansion of Mart-1 and Flu antigen specific tetramer positive cells in IVCs with total PBL or purified CD8+ cells. Figure 1a = Mart-1 and Flu specific tetramer positive CD8+ cells from PBL IVC. Left panel shows baseline antigen specific cells: Top is Mart-1, bottom is Flu. Right panel shows expanded antigen specific cells: Top is Mart-1, bottom is Flu. Figure 1b = Mart-1 and Flu specific tetramer positive CD8+ cells in IVC with purified CD8+ cells. Left panel shows baseline antigen specific cells: Top is Mart-1, bottom is Flu. Right panel shows expanded antigen specific cells: top is Mart-1, bottom is Flu. Figure 1c = Specific cytotoxic effect of expanded CTL in IVCs. Assays were done in 51Chromium release using T2 cells pulsed with different peptides as target cells. M3, a nine amino acid peptide binding with A2 is used as control peptide. Figure 1d = Specificity in IFN-γ secretion by expanded CTL in IVCs as determined by ELISA with supernatant collected 24 h after re-stimulation of the expanded cells.
View Figure 1
Figure 1: Expansion of Mart-1 and Flu antigen specific tetramer positive cells in IVCs with total PBL or purified CD8+ cells. Figure 1a = Mart-1 and Flu specific tetramer positive CD8+ cells from PBL IVC. Left panel shows baseline antigen specific cells: Top is Mart-1, bottom is Flu. Right panel shows expanded antigen specific cells: Top is Mart-1, bottom is Flu. Figure 1b = Mart-1 and Flu specific tetramer positive CD8+ cells in IVC with purified CD8+ cells. Left panel shows baseline antigen specific cells: Top is Mart-1, bottom is Flu. Right panel shows expanded antigen specific cells: top is Mart-1, bottom is Flu. Figure 1c = Specific cytotoxic effect of expanded CTL in IVCs. Assays were done in 51Chromium release using T2 cells pulsed with different peptides as target cells. M3, a nine amino acid peptide binding with A2 is used as control peptide. Figure 1d = Specificity in IFN-γ secretion by expanded CTL in IVCs as determined by ELISA with supernatant collected 24 h after re-stimulation of the expanded cells.
View Figure 1
Table 1: Cytotoxic effect of CD8+ cells expanded in PBL IVC.
Functional characterization of expanded CD8+ Mart-1 antigen specific cells expanded in PBL + autologous tumor IVCs by cytotoxicity assays using autologous and allogeneic tumor cells as targets.
View Table 1
We noticed that Mart-1specific or tumor specific CD8+ CTLs in PBL IVC disappeared within two to three weeks despite repeated stimulations with appropriate stimuli, see Table 2. Whereas the Flu specific CTL generated with total PBL continued to expand and maintained their functions (data not shown). The above observation of loss of Mart-1 specific CTL or tumor specific CTL with total PBL might be due to a significant difference between the phenotypic and functional properties of the expanding CD4+ cells, the induced Treg cells, in those co-cultures with PBL. This observation led us to characterize those induced Tregs in culture.
Table 2: Fate of induced CD8+ CTLs generated with PBL in cultures with autologous tumors or Mart-1 peptide pulsed DC.
"+" = Cytokine secreted more than 50 pg and cytotoxic effect more than 20% killing of the target cells with highest Effector cells: target cells. "++" = Cytokine secreted more than 100 pg/ml and cytotoxic effect more than 40% with highest Effector: target cells. "-" = No cytokine or no cytotoxic effect.
View Table 2
CD4+ cells from the PBL IVCs with different conditions were purified by positive selection with magnetic beads. These CD4+ cells were then re-stimulated with plate bound anti-CD3 or with autologous DC (responder: stimulator = 10:1). Figure 2 describes phenotypic and functional profile of CD4+ cells taken from the PBL cultures by intracellular staining. Figure 2a describe the phenotype of such Tregs induced under different conditions. We found significantly higher levels of IL-10 synthesized by CD4+ cells taken from Mart-1 IVC or autologous tumor IVC. The percentage of CD4+ and FoxP3+ cells varied from 2.1 to 3.2 in freshly isolated total PBL (data not shown). The different types of CD4+ Tregs are from different culture conditions as follows A = CD4+CD25+ cells isolated from PBL IVC with Mart 1peptide pulsed auto- DC. B = CD4+CD25+ cells isolated from PBL IVC with Flu peptide pulsed auto-DC. C = CD4+CD25+ cells isolated from PBL IVC with autologous tumor.
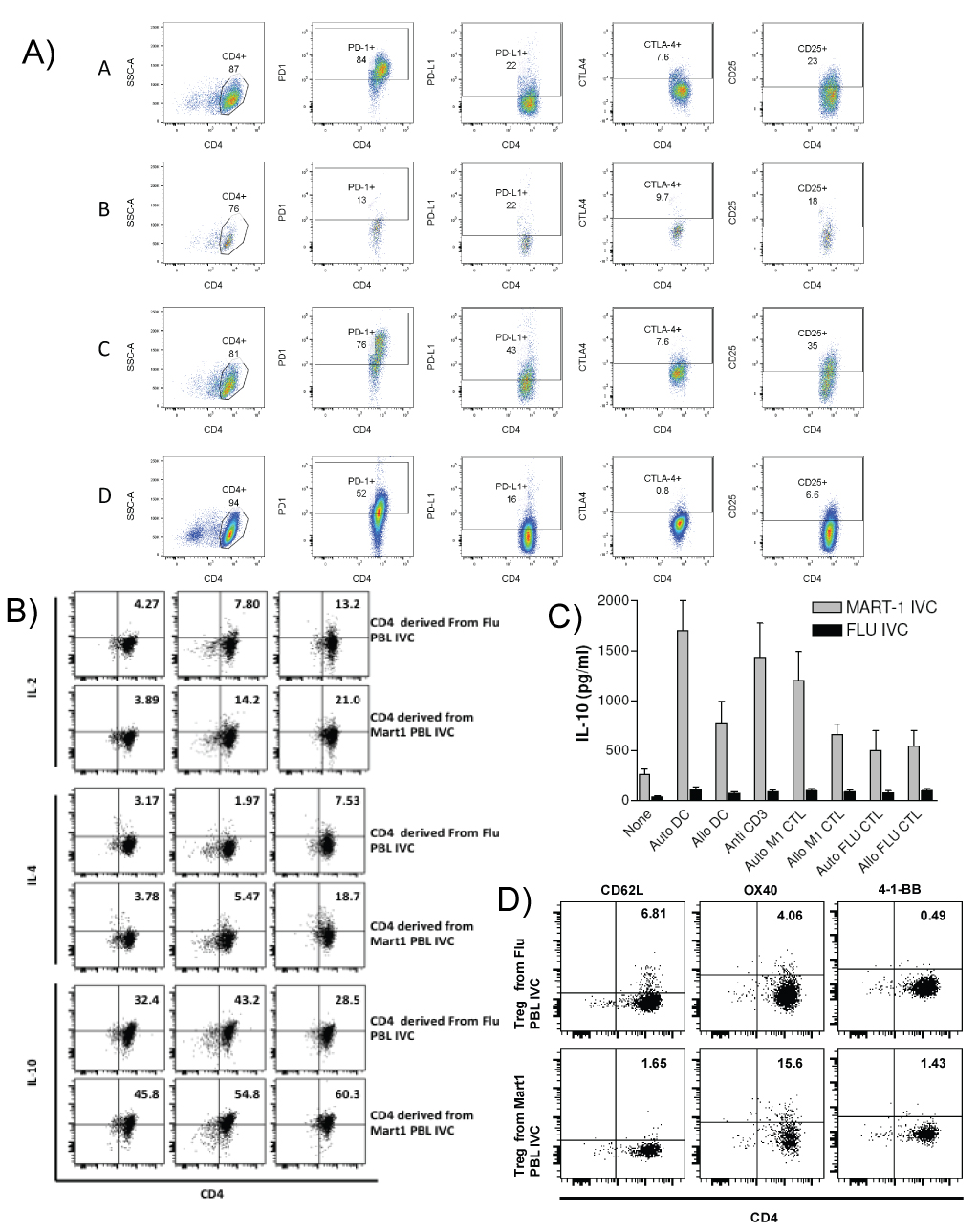 Figure 2: Phenotypic and functional characterization of CD4+ cells isolated from Mart-1 and Flu PBL IVC. CD4+ cells were isolated from Mart-1 and Flu antigen specific PBL IVCs after 3rd stimulation. Phenotypic analysis of the purified CD4+ cells from PBL IVC were performed by staining these cells with CD4+, CD25 and Foxp3 and CD62L, OX40 and 4-1-BB. Figure 2a = Phenotypic analysis of CD4+ isolated from Mart-1 and Flu antigen stimulated PBL IVC. The different types of CD4+ Tregs are from different culture conditions as follows: A = CD4+CD25+ cells isolated from PBL IVC with Mart 1peptide pulsed auto- DC. B = CD4+CD25+ cells isolated from PBL IVC with Flu peptide pulsed auto-DC. C = CD4+CD25+ cells isolated from PBL IVC with autologous tumor D = CD4+CD25+ generated with purified CD4+CD25- cells in culture with anti CD3+anti CD28 +IL-2. Figure 2b = Intracellular cytokine production by Treg cells after stimulation with anti-CD3 antibody. Number of IL-2, IL-4 and IL-10 producing cells were analyzed by FACS. Figure 2c = Tregs from PBL IVCs A & B were further compared for level of IL-10 production after re-stimulation. Figure 2d = Comparative analysis of CD62L, OX and 41BB expression by two different Tregs isolated from PBL IVCs A &B.
View Figure 2
Figure 2: Phenotypic and functional characterization of CD4+ cells isolated from Mart-1 and Flu PBL IVC. CD4+ cells were isolated from Mart-1 and Flu antigen specific PBL IVCs after 3rd stimulation. Phenotypic analysis of the purified CD4+ cells from PBL IVC were performed by staining these cells with CD4+, CD25 and Foxp3 and CD62L, OX40 and 4-1-BB. Figure 2a = Phenotypic analysis of CD4+ isolated from Mart-1 and Flu antigen stimulated PBL IVC. The different types of CD4+ Tregs are from different culture conditions as follows: A = CD4+CD25+ cells isolated from PBL IVC with Mart 1peptide pulsed auto- DC. B = CD4+CD25+ cells isolated from PBL IVC with Flu peptide pulsed auto-DC. C = CD4+CD25+ cells isolated from PBL IVC with autologous tumor D = CD4+CD25+ generated with purified CD4+CD25- cells in culture with anti CD3+anti CD28 +IL-2. Figure 2b = Intracellular cytokine production by Treg cells after stimulation with anti-CD3 antibody. Number of IL-2, IL-4 and IL-10 producing cells were analyzed by FACS. Figure 2c = Tregs from PBL IVCs A & B were further compared for level of IL-10 production after re-stimulation. Figure 2d = Comparative analysis of CD62L, OX and 41BB expression by two different Tregs isolated from PBL IVCs A &B.
View Figure 2
D = CD4+CD25+ generated with purified CD4+CD25- cells in culture with anti CD3+anti CD28 +IL-2 as a conventional way of generating Tregs in culture. Figure 2b shows the intracellular Il-10 production by the Tregs. Here the effects of only two different Treg cells are shown for comparison between self (Mart-1) and non-self (Flu) antigens. Figure 2c shows the IL-10 production profile by these cells by cytokine ELISA. When we analyzed for the expression of CD62L, homing receptor and for central memory on these CD4+ cells, we found CD62L is much higher in CD4+ cells taken from Flu IVCs Figure 2d. While comparing the CD4+ cells isolated from two different IVCs we found 3 times more of CD4+ cells from Mart-1 IVC, expressing OX40. We did not detect any significant difference in expression of 4-1BB between these CD4+ cells taken from two different IVCs (Figure 2d).
We then tested these induced Treg cells in separate CTL generation assays for their functional properties. Table 3 shows the results of functional properties of the Tregs isolated from different conditions of co-cultures. Here we present data with six HLA A2+ patients (UC 7 to UC 12). We used two different types of induced Treg cells (A and B, for the comparison between self and non-self) isolated from different culture conditions as shown in Figure 2a. In the assays we stimulated purified CD8+ Tells from each HLA A2 + patient, with respective autologous DC pulsed with Mart-1 peptide (AAGIGILTV) in the presence and absence of respective autologous Tregs isolated from Mart-1 PBL IVC or Flu PBL IVC as described in the Figure 2. It can be seen that Tregs induced from Mart-1 or tumor cell cultures (data with Tregs from C & D not sown), blocked the CTL induction in the cultures Table 2. Cytotoxicity assays were done with the CTLs, generated with CD8+ cells from individual patient, using T2 cells (gift from Dr. Creswell of Yale University) [12] pulsed with Mart-1 peptide as target cells. Percent of target cell killing were measured in a standard chromium release assay as described in materials methods. Killing assays were done in triplicate for each case and expressed as average killing ± SD. In all the six cases significant blocking of CTL induction were observed with Treg A and not with TregB. Reasonable blocking of CTL generation was also observed with Treg C and Treg D (data not shown).
Table 3: Effect of induced Tregs on CTL generation with purified CD8+ Cells.
Functional characterization of expanded CD4+ Tregs in separate in vitro Mart-1 specific CTL generation assays with purified CD8+ cells stimulated with autologous DCs pulsed with Mart-1 peptide.
View Table 3
The major population of Foxp3+ Treg is generated in the thymus, now known as tTreg [1-3]. In the periphery Tregs are generated from T-conventional (Tconv) cells. Such cell populations are termed as peripherally derived Tregs (pTregs) [1,14]. The mechanism of induction of these pTreg cells in the periphery is not known [1,4,7]. We tried to address the important issues regarding the induction and functions of these pTreg and/or iTreg subpopulations in an in vitro set up (as seen in the schematic diagram, Figure 3). A detailed characterization of these subpopulations has important clinical implications in cellular immunotherapy for cancer [1-4,13,14]. Tumor associated antigen specific CD8+ CTL have shown considerable promise in translational tumor immunotherapy [15,16]. In this context our in vitro induction of CTL generation and corresponding expansion of T regs in culture are important. Variety of intrinsic regulatory processes within T cells as well as a number of microenvironment-based extrinsic mechanisms have turned out to be serious impediments in achieving robust and sustained outcomes of CTL mediated immunotherapy for cancer [11]. Polyclonal tTregs might specifically target trafficking pathways and not allowing immunity to develop in lymphoid organs. That could be the reason for the existence of tumor specific CD8+ CTL in tumor tissues. Under such conditions a specific Treg cells needed to be induced from conventional CD4+CD25- T cells to work against those auto-reactive T cells that are generated in the periphery [4,5,14,17,18].
 Figure 3: Induction of pTregs in tumor microenvironment and elimination of tumor specific CTL response.
View Figure 3
Figure 3: Induction of pTregs in tumor microenvironment and elimination of tumor specific CTL response.
View Figure 3
We have made an attempt to understand the functional properties of the CD4+CD25+ cells induced from CD4+ CD25- cells from the co-cultures and we found that indeed those Treg cells suppress the CTL generation (Figure 2 and Table 2). This induced Tregs from culture secrete IL-10, that dampens the antigen presenting ability of DC (Figure 3). This finding is similar to previous reports by other laboratories and us [5,17,19,20].
Naive and activated T cells are known to express different adhesion molecules and are thought to exhibit different migratory patterns that result from their expression of discrete adhesion molecules. Two known adhesion molecules that have been associated with differentiating the naive and activated/memory T cells, are CD62L (L-selectin) and CD44 (H-CAM) [21,22]. In our in vitro CD8+ CTL generation experiments against Mart-1 and Flu we found a marked difference in lymphoid homing receptor expression. It is known that naive T cells express a CD62Lhi CD44lo phenotype, whereas memory T cells exhibit a CD62Llo CD44hi phenotype [20-22]. In this regards increased number of CD62L expressing CD4+ cells from Flu IVC, could be indicative of memory cells against Flu matrix protein derived antigens, as most of us are exposed to Flu antigen at least once in life time where as Mart-1 being a self antigen no such thing as memory cells were seen (Figure 2d).
The number of CD4+ cells that expressed CD25 or Foxp3 from these cultures is not really comparable (Figure 2a). This can be explained by the fact that under different conditions of IVCs, or in vivo tumor microenvironment. Where CD4+ cells have already seen the autologous tumor antigen or peptide epitopes. So in either case the CD4+ T cells, in vitro or in vivo, behave as T regulatory cells to prevent tumor (auto)destruction.
The host's capacity to respond to a given tumor antigen measured in assays with T cells derived from the circulation does not necessarily reflect the host's immunological responsiveness, in situ, at the tumor sites. Microscopic studies of tumors for infiltrating lymphocytes as well as immune-histochemical examination of tumor explants, carried out with outcome analyses, have provided useful information. Similarly, in vitro phenotypic and functional analyses of T cells derived from peripheral blood have also provided useful information on the nature of the host immune responses to tumors. Our findings suggest that in vitro functional T cell immune characteristics could be examined in patients diagnosed with metastatic melanoma along with their status of regulatory activity. Examination of these characteristics with larger number of patients might provide predictive marker for immune intervention.
This work was supported by a grant from CRC, MO 1RR06192, UConn Health and Richard and Jane Lublin (UPH).