Anti-phospholipid syndrome (APS) or Hughes syndrome is considered as one of the most common autoimmune disorders that present as a hypercoagulable state accompanied by laboratory findings of anti-phospholipid antibodies (aPLs). The development of a previously diagnosed primary APS with no clinical and diagnostic evidence of other autoimmune diseases followed by systemic lupus erythematosus (SLE) is a rare occurrence. SLE may develop years after the onset of the primary APS. A 30-year-old Filipino female initially presented with thromboembolic symptoms (massive pulmonary embolism, recurrent pregnancy losses, and deep venous thrombosis) and tested positive for anti-phospholipid antibodies with negative findings for SLE. She developed joint pains, alopecia, and myalgia accompanied by thrombocytopenia, neutropenia, and hemolytic anemia eight years later, which resulted in a diagnosis of an SLE with secondary APS. The patient was initially treated with warfarin but was shifted to dabigatran due to problems in monitoring. During the succeeding years, there were recurrent episodes of pulmonary embolism despite antiplatelet, heparin, and steroid use, which resulted in the development of chronic thromboembolic pulmonary hypertension (CTEPH). The patient continued with her anticoagulation and underwent pulmonary endarterectomy. The mainstay of treatment for thromboembolism in APS requires anticoagulation with warfarin or heparin and aspirin, but other alternatives such as direct oral anticoagulants (DOACs) should be considered, especially in extenuating circumstances.
Anti-phospholipid syndrome, Systemic lupus erythematosus, Anticoagulation
Antiphospholipid Syndrome (APS) or Hughes syndrome is an autoimmune disorder that presents clinically as thrombosis in the arterial and venous circulation and recurrent pregnancy losses accompanied by evidence of anti-phospholipid antibodies [1]. The development of anti-phospholipid antibodies is found in 1-5% of the general population, but only 5 in 100,000 persons will develop APS [2-4]. APS can be classified as primary if the disease is not in association with any other autoimmune disease, secondary if APS is secondary to another autoimmune disease, or catastrophic in which there is the development of thrombosis leading to multi-organ failure in a span of fewer than one week [5]. There may be rare instances in which an initial diagnosis of a primary APS will progress into a secondary APS after several years [6]. The determination of whether APS is primary or secondary based on symptomatology is difficult as they may share similar clinical findings. However, there may be some clues clinically and diagnostically to determine each classification.
This case presented a 30-year-old Filipino female with signs and symptoms of recurrent thromboembolism (massive pulmonary embolism, recurrent pregnancy losses, and deep venous thrombosis) for the past 11 years associated with high titers of anti-phospholipid antibodies (aPLs). In the interim, she developed a catastrophic type of APS and later on developed chronic thromboembolic pulmonary hypertension (CTEPH) despite adequate anticoagulation. She was diagnosed with systemic lupus erythematosus (SLE) 8 years later due to persistently high ANA titers, joint pains, alopecia, thrombocytopenia, neutropenia, and hemolytic anemia, and chronic urticaria.
The complexity of the clinical manifestations and management of this patient may contribute to the recognition and understanding of APS in association with SLE, which made this case reportable.
A 30-year-old Filipino female presented with a chief complaint of dyspnea. History started 11 years before admission when she experienced right leg swelling with occasional pain on exertion. She sought consultation with a cardiologist and was given unrecalled antiplatelet with temporary relief (Figure 1). Multiple consults were done in the next two years due to the recurrence of the leg swelling, and work up revealed deep venous thrombosis. The patient was started on warfarin, which provided relief.
 Figure 1: History timeline of the clinical history of a 30-year-old Filipino female with a chief complaint of dyspnea.
View Figure 1
Figure 1: History timeline of the clinical history of a 30-year-old Filipino female with a chief complaint of dyspnea.
View Figure 1
Eight years before admission, she experienced a sudden onset of dyspnea, accompanied with non-radiating chest and flank pain. She was brought to a local hospital wherein pain management was done and was subsequently discharged. Two years later, she had an abortion at ten weeks AOG.
Hypercoagulability was one of the differentials; thus, she was referred to an immunologist for workup. She was found out to be strongly positive for lupus anticoagulants (PTT-LA, Kaolin clotting time, Dilute Russel viper venom time) and the levels of her cardiolipin and B2 glycoprotein antibodies were moderate to high (Table 1). On her second pregnancy, she was given aspirin 80 mg once a day, and a low molecular weight heparin (LMWH) injection (tinzaparin sodium 4500 IU/mL, SQ OD). At the 24th week of gestation, she was admitted for sudden onset of dyspnea. Work up was done, and was found out to have a pulmonary embolism. She was immediately placed on a heparin drip, but it took a while before her dyspnea was relieved, and her fetus died in utero a few days later. Her heparin was later shifted to warfarin, and she went home to the province.
Table 1: Antiphospholipid Panel Result dated 2012. View Table 1
During the treatment with warfarin, the patient was not able to reach the therapeutic levels (INR of 2-3) due to inaccessibility for diagnostic monitoring. Thus, the patient was started on dabigatran 150 mg bid instead. Over the next three years, she had recurrent mild dyspnea on exertion.
Five years prior to admission, the persistence of the dyspnea was noted with occasional symptoms of chest pain and flank pain. Elevated pulmonary pressures on echocardiography were likewise noted. Further testing through computed tomography (CT) and transesophageal echocardiography were consistent with complete occlusion of the right main pulmonary artery and elevated pulmonary pressures at 83 mmHg with a dilated right ventricle (Figure 2 and Figure 3). The patient was started on sildenafil 100 mg once a day. Since the patient resided in the province at that time, low molecular weight heparin (LMWH) was too expensive and difficult to procure, and the warfarin therapeutic level was difficult to monitor; hence dabigatran was continued. The patient also developed chronic urticaria and was treated with levocetirizine and later, with Bilastine. Her CBC showed normal hemoglobin and WBC, but with mild thrombocytopenia (Table 2). Her repeat APS panel showed moderate to high levels of aPLs; her ANA titer was high with normal anti-dsDNA and C3. Since mild thrombocytopenia can be part of the APS, therefore SLE was not considered at that time.
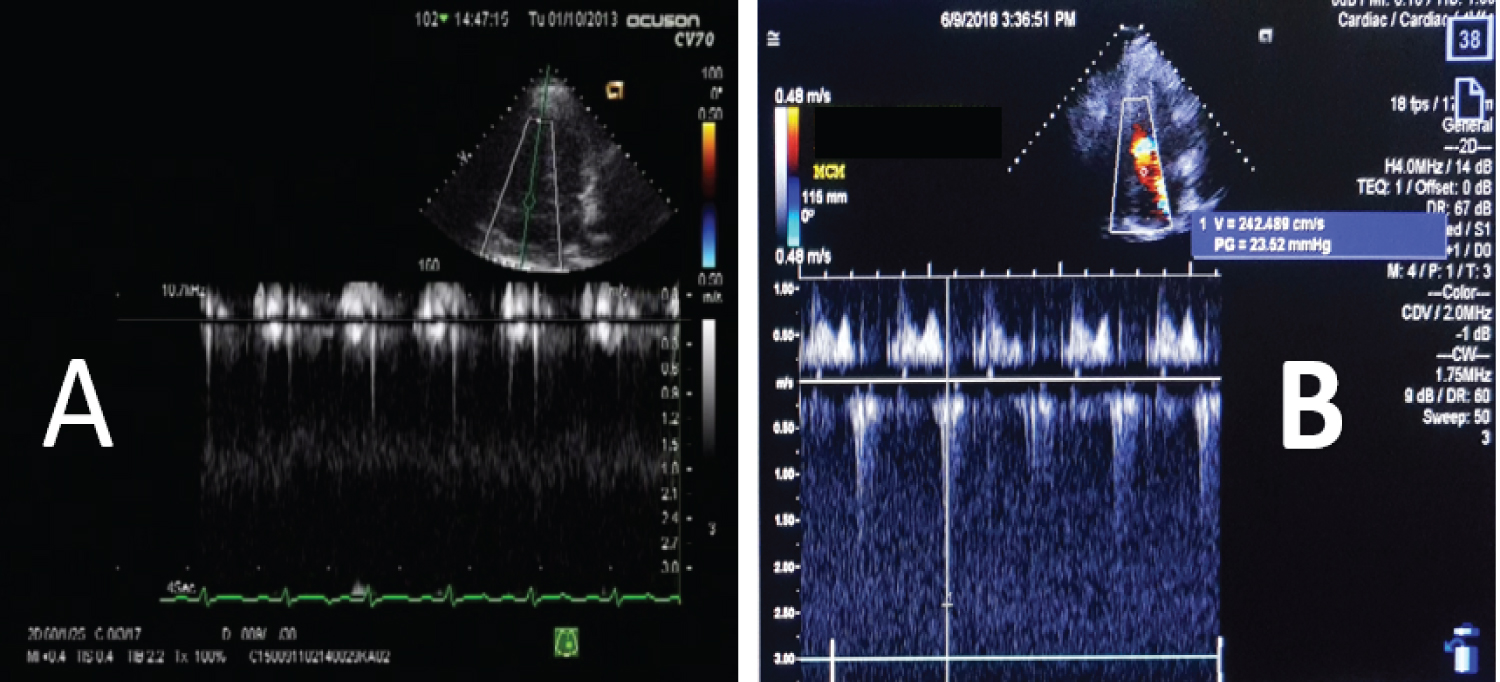 Figure 2: (A) Echocardiography done preoperatively showing increase in pulmonary pressure; (B) Echocardiography done postoperatively with noted improvement in the pulmonary pressure.
View Figure 2
Figure 2: (A) Echocardiography done preoperatively showing increase in pulmonary pressure; (B) Echocardiography done postoperatively with noted improvement in the pulmonary pressure.
View Figure 2
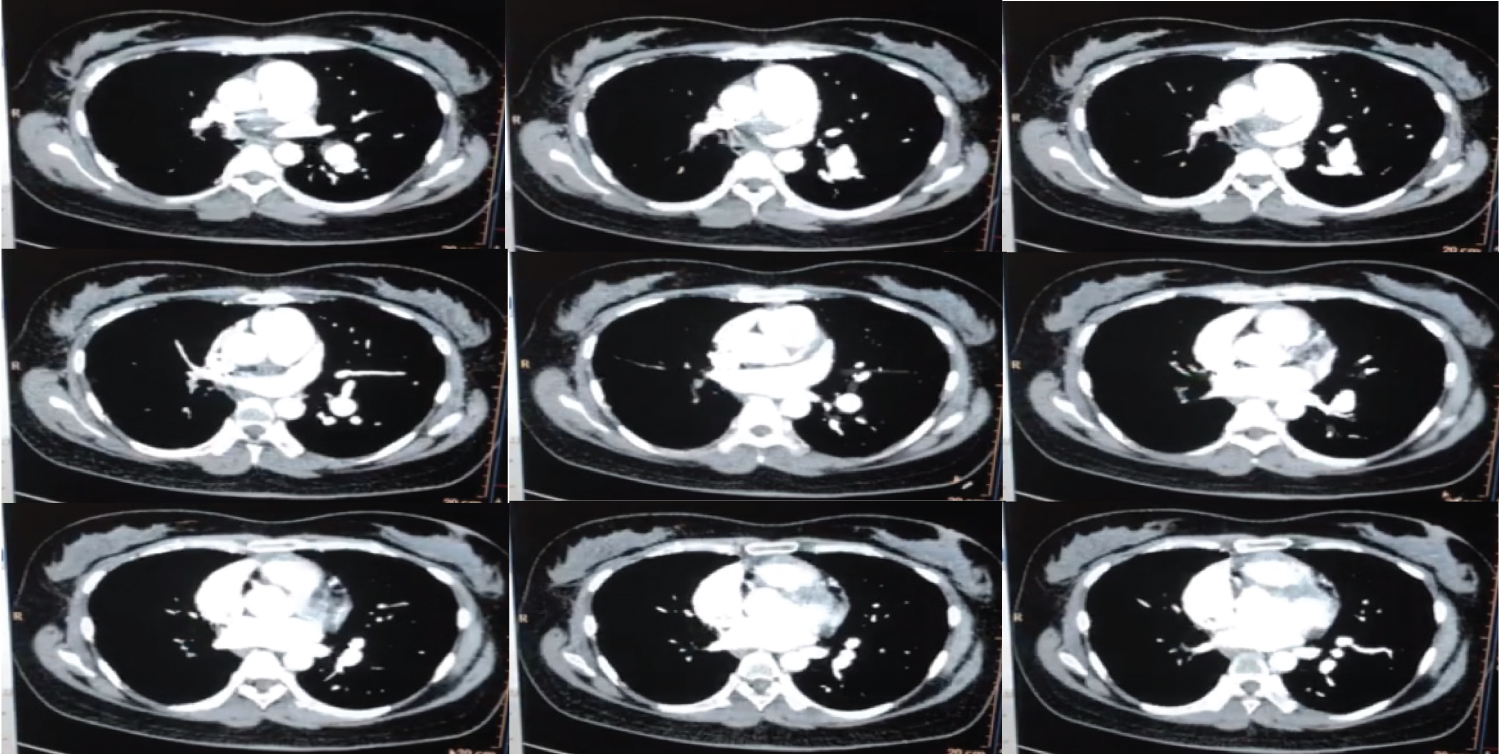 Figure 3: Chest CT findings revealing a thrombus on the right pulmonary artery and intravascular thrombi in the segmental branches. There is also note of absence of the arterial opacification in the right lung.
View Figure 3
Figure 3: Chest CT findings revealing a thrombus on the right pulmonary artery and intravascular thrombi in the segmental branches. There is also note of absence of the arterial opacification in the right lung.
View Figure 3
Table 2: Complete blood count trends during the course of the illness. View Table 2
Two years before admission, moderate thrombocytopenia (84 × 109/L - 112 × 109/L) was already noted, followed by the patient's complaints of alopecia, joint pains, muscle pains, along with the persistent dyspnea and chest pains. Laboratory results revealed anemia of 86 g/L, elevated titers of ANA, low C3, and a positive Coomb's test, among others (Table 3). Although her anti-dsDNA remained negative, she was diagnosed with SLE. Hydroxychloroquine 200 mg was added to her regimen, but she developed generalized morbilliform pruritic rashes after a few days; hence it was discontinued. She was placed on long term oral prednisone by the rheumatologist.
Table 3: Summary of laboratory results during 2 years prior to admission. View Table 3
In the interim, the patient was on constant follow up with her physicians and was appraised for possible surgical intervention. However, the patient initially was not amenable to such treatment options due to her mild symptoms of dyspnea and pain. Despite the ongoing steroids, thrombocytopenia persisted and became severe.
Seven months before admission, a repeat chest CT scan with contrast revealed complete occlusion of the right main pulmonary artery and main secondary branches with chronic thrombosis. There was a progression of the patient's dyspnea, which made her reconsider the option of surgical removal of the thrombus.
One month before admission, the patient was admitted to another institution because of severe thrombocytopenia (44 × 109/L). She was referred to a hematologist to correct the thrombocytopenia prior to cardiac catheterization and eventual thrombectomy and pulmonary endarterectomy. Despite on high dose steroids and platelet concentrate transfusions, the thrombocytopenia remained below 50 × 109/L. Intravenous gamma globulin (IVIg) was given at 30g infusion (the patient could not afford the 1g per kg per dose), and the platelet rose to 109 × 109/L - 118 × 109/L. She was then able to undergo right cardiac catheterization, which revealed severe pulmonary hypertension (Figure 4). The patient was advised for thrombectomy and pulmonary endarterectomy and was then admitted for the procedure.
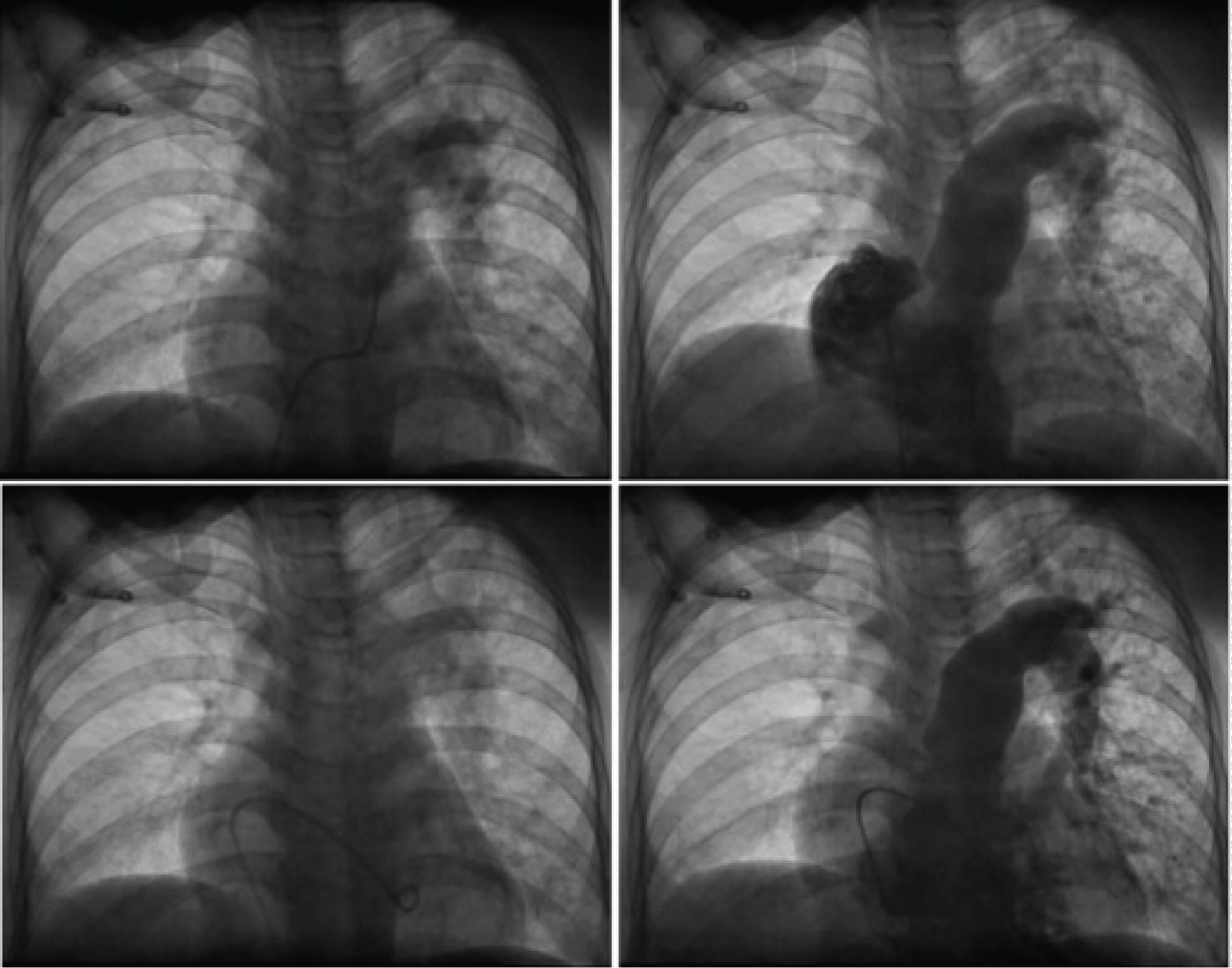 Figure 4: Right cardiac catheterization revealing severe pulmonary hypertension, dilated main pulmonary artery and elevated right and left end diastolic pressure.
View Figure 4
Figure 4: Right cardiac catheterization revealing severe pulmonary hypertension, dilated main pulmonary artery and elevated right and left end diastolic pressure.
View Figure 4
The patient was previously treated for syphilis due to a positive Venereal Disease Research Laboratory (VDRL) result. She was also previously diagnosed with chronic urticaria and allergic rhinitis. The patient was a gravida two para 0 abortion 2 with regular menstrual interval and duration. She was a non-smoker, a nonalcoholic beverage drinker, and worked as a firefighter.
On admission, the patient was prepared for the procedure of thrombectomy, open-heart surgery, cardiopulmonary bypass with circulatory arrest. The 12-lead ECG showed right ventricular hypertrophy and anteroseptal wall ischemia (Figure 5). There was a recurrence of thrombocytopenia (104 × 109/L) and was corrected with another intravenous immunoglobulin infusion.
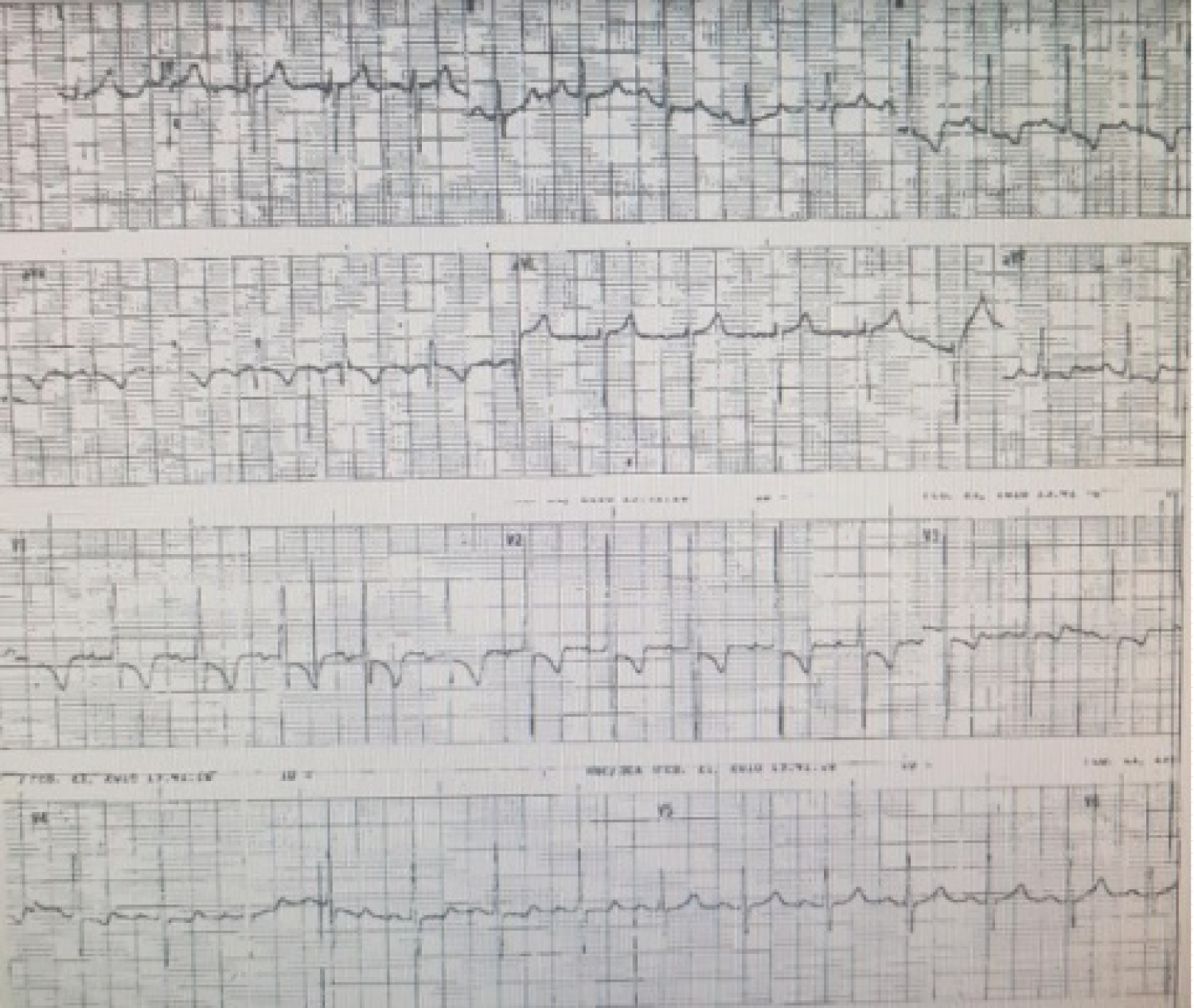 Figure 5: 12-lead electrocardiogram revealed right ventricular hypertrophy and right atrial enlargement and anterolateral wall ischemia.
View Figure 5
Figure 5: 12-lead electrocardiogram revealed right ventricular hypertrophy and right atrial enlargement and anterolateral wall ischemia.
View Figure 5
The patient underwent the procedure on the 3rd day of admission. Intraoperatively, a firm doughy round clot, was removed from the main pulmonary artery (Figure 6). Postoperatively, the patient was transferred immediately to the recovery room and later to the surgical intensive care unit (ICU) after three days. The patient's course in the surgical ICU was unremarkable. She was extubated from the ventilator and was resumed on oral medications on the post-operative day.
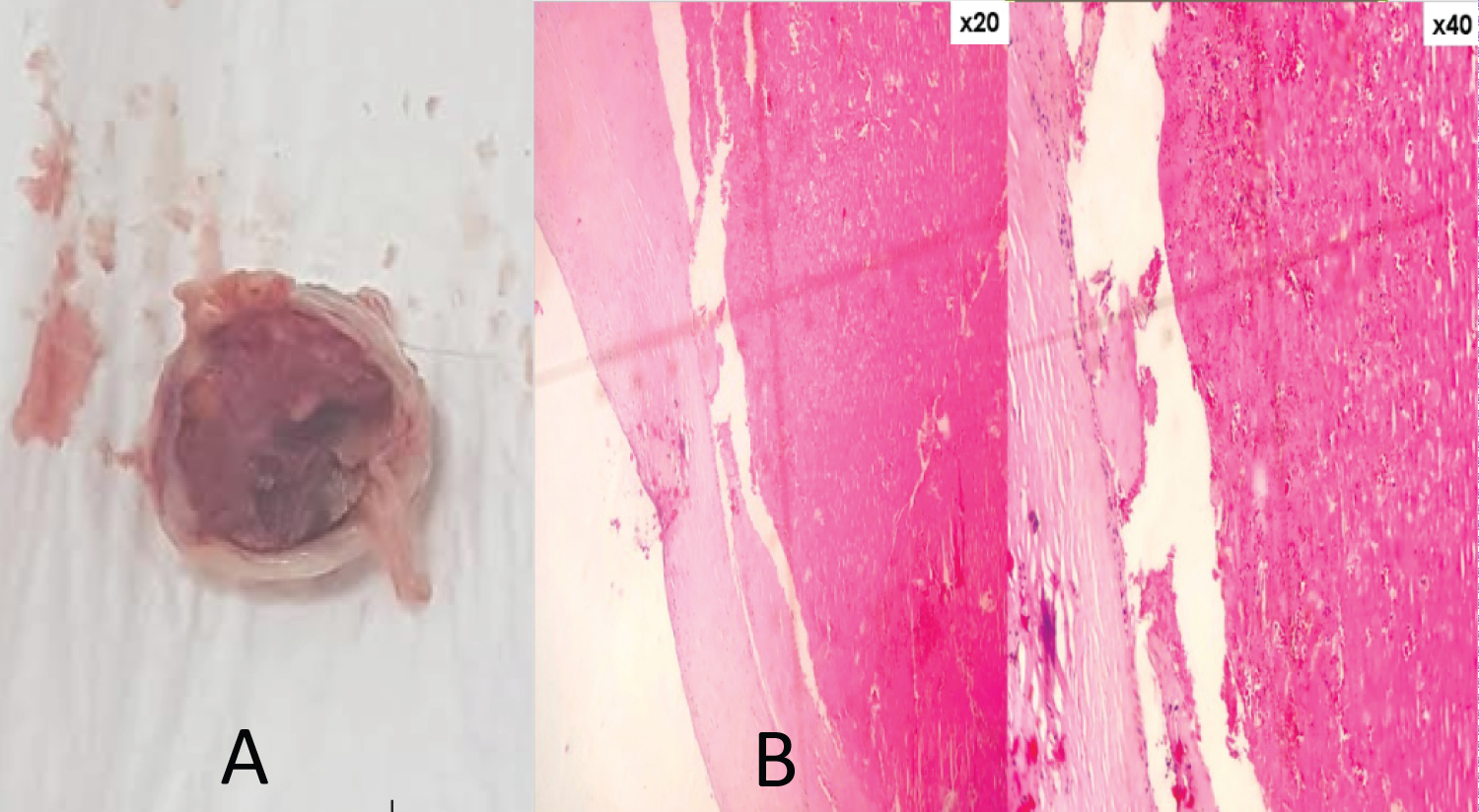 Figure 6: (A) Thrombus removed from the right main pulmonary artery measuring approximately 2.3 cm in largest diameter with (B) Histopathologic findings consistent with a thrombus.
View Figure 6
Figure 6: (A) Thrombus removed from the right main pulmonary artery measuring approximately 2.3 cm in largest diameter with (B) Histopathologic findings consistent with a thrombus.
View Figure 6
Repeat echocardiography postoperatively revealed mild to moderate pulmonary hypertension with pulmonary regurgitation, a right ventricular ejection fraction of 40-42%, and left ventricular ejection fraction of 66% (Figure 2).
She was transferred back to the ward on the 7th post-operative day. No significant events occurred thereafter, and the patient was discharged on the 12th hospital day. On follow up, the histopathology of the specimen was consistent with a thrombus (Figure 6).
The diagnosis of APS is usually suspected in young female patients presenting with recurrent thromboses pregnancy losses combined with laboratory findings confirming elevated levels of anti-phospholipid antibodies [1]. The Sapporo criteria in APS entails at least one clinical and one laboratory findings repeated at least 12 weeks apart to confirm the diagnosis of APS. The rationale for repeating the panel after 12 weeks is to be able to eliminate other causes of a phospholipid antibody reactivity secondary to sepsis or other blood-related disorders such as acute disseminated intravascular dissemination (DIC), thrombotic thrombocytopenia, hemolytic uremic syndrome, and others [1,2]. However, the inability to fulfil the criteria does not hinder the clinician from diagnosing a patient with APS, especially if clinically apparent since the criteria only serve as a guide to the diagnosis and not meant to be followed strictly [1]. In comparison, the diagnosis of SLE makes use of the American Rheumatism Association (ARA) criteria that confirm the diagnosis of SLE if 4 out of 11 symptoms are present and the Systemic Lupus International Collaborating Clinics (SLICC) criteria, which needs at least one clinical and one immunologic criterion to clinch the diagnosis of SLE [7].
The occurrence of a primary APS is more common than APS accompanied by other autoimmune diseases [1]. In a study done in Singapore, the prevalence of primary and secondary APS in Singapore was 61% and 39%, respectively [6]. In comparison, secondary APS is found in 10% of cases with SLE worldwide. Other studies suggest that the SLE with secondary APS is seen in 22.4% of cases [8].
The development of a primary APS into a secondary APS with SLE is an uncommon occurrence. To date, written literature has cited 30 cases of patients who initially presented with a primary APS who developed into SLE [9].
The patient, in this case, presented with an 8-year interval from primary APS to the development of secondary APS secondary to SLE. Different studies have shown that the progression from a primary to a secondary APS with SLE will take months to years. One case reported that the evolution of a primary APS into SLE is two years from the onset of thrombotic events, while another study done in Colombia revealed an interval of 1-7 years [10,11].
The presence of thrombocytopenia can be a clinical manifestation of APS that is not included in the Sapporo criteria, and may also be a hematologic manifestation of SLE [11]. In the patient's case, thrombocytopenia was initially mild, and the progression to severe coincided with the appearance of the other manifestations of SLE.
The primary mechanism in the pathophysiology in APS requires binding of the aPLs to the B2 glycoprotein I receptors leading to a cascade of events promoting thrombosis. Initially, the anti-phospholipid antibodies are produced by the B cells and bind to the different B2 glycoprotein I receptors found in the endothelium and platelets and may also lead to the activation of the complement pathway [1]. The interaction with the antibodies and endothelial cells receptors will lead to a decrease in the production of nitric oxide, thus promoting thrombosis [12,13]. This interaction induces the production of thromboxane B2, leads to an increase in thrombotic events [2,13]. Though there is a small group of healthy patients who turn positive for the antibodies, the 2-hit hypothesis is suggested for APS to develop. The 1st hit describes the interaction with the aPLs accompanied by and endothelial dysfunction, while the 2nd hit results in the increased activity of the B2- glycoprotein receptors induced by inflammation [13].
The occurrence of miscarriage is a fundamental obstetric symptom in APS. The mechanism of APS in a patient presenting with recurrent fetal losses is secondary to the interaction of the anti-B2 glycoprotein I antibodies to the exposed phospholipids of the trophoblast during pregnancy [13]. Furthermore, this interaction leads to the inhibition of the implantation of the trophoblast. These anti-phospholipid antibodies then interact with the placental wall to cause blood clots that may cause placental disruption and decreased oxygenation to the fetus leading to restricted growth and abortion [1,13].
The venous thrombosis presented as an 11-year history of recurrent deep vein thrombosis and pulmonary embolism in this patient is the most common presenting symptom in patients with APS [5,14]. The elevated pulmonary pressures and occluded main pulmonary artery, as seen on imaging, were consistent with chronic thromboembolic pulmonary hypertension (CTEPH). CTEPH is characterized by the presence of a fibrotic thrombus secondary to inadequate anticoagulation and elevated pulmonary pressures of > 25 mmgHg on right cardiac catheterization [15]. The patient presented initially with a sudden onset of dyspnea eight years before admission and had resolution of the dyspnea and chest pain. The clinical course was consistent with the initial presentation of CTEPH, wherein signs and symptoms of pulmonary embolism were initially noted, followed by a period of being asymptomatic or known as the 'honeymoon period' [15].
It is also worth noting that the patient developed chronic urticaria during the course of the illness. In a meta-analysis, 22% of patients with SLE may present with chronic urticaria, and the pathophysiology is mainly secondary to an immune complex deposition [16]. Furthermore, the same study suggested that the presence of chronic urticaria in patients may be a precursor of and maybe a sign of a higher risk of developing SLE [16].
The patient's symptoms of dyspnea, chest pain, miscarriage at 24 weeks AOG that happened eight years before admission may be a manifestation of a catastrophic APS. The onset of the respiratory symptoms was sudden, accompanied by different thrombotic episodes, namely pulmonary embolism, deep venous thrombosis, and miscarriage despite aggressive anticoagulation that fulfilled the criteria [5].
Clinically, distinguishing primary from secondary APS is difficult as they have similar clinical manifestations [12]. However, constant clinical monitoring and repeated diagnostic testing were essential in determining the progression from primary APS to SLE with secondary APS. Other diagnostic parameters can help determine the development of SLE from an initial primary APS. In this case, laboratory findings of a positive Coomb's test with anemia, leukopenia, and thrombocytopenia were noted. Studies suggest that the presence of these hematologic findings would indicate the development of SLE from a primary APS [11,12]. Furthermore, hemolytic anemia is usually seen in SLE with APS rather than a primary APS [12].
The standard of care in the medical management of venous thromboembolism in APS patients is the use of heparin or the use of Vitamin K antagonists such as warfarin, while the management in pregnant patients with APS makes use of aspirin and heparin [17,18]. In this case, the patient was started on warfarin, but due to the inability to monitor and achieve therapeutic levels, the use of direct oral anticoagulants (DOACs) to treat the recurrent thromboses was considered. The advent of DOACs was used primarily for valvular heart diseases, and little is still known on its efficacy of anticoagulation in patients with recurrent thrombosis in APS. Though no concrete studies are currently available to support the use of DOACs in secondary prevention of thrombosis in APS [19], the alternative use of dabigatran in this patient was done since she has been experiencing multiple thrombotic episodes and was unable to maintain an optimal level of anticoagulation with warfarin and had difficulty in procuring an LMWH as well. Four cases that used Rivaroxaban as thromboprophylaxis in patients with APS showed no recurrence in thrombosis when treated for six months with no significant differences than warfarin [20]. A similar study and case presented with triple-positive anti-phospholipid antibodies and poorly controlled INR levels provided similar evidence that the use of DOACs (Rivaroxaban) may be a good substitute for the treatment of recurrent thrombosis in APS [18].
The mainstay of treatment for CTEPH remains to be pulmonary endarterectomy and is noted to be lifesaving [12,21]. The procedure will directly relieve and decrease the pulmonary pressure, thus relieving the symptoms of chest pain and dyspnea. The patient improved postoperatively after the pulmonary endarterectomy, as evidenced by the clinical improvement in symptoms and decreased pulmonary pressures on echocardiography.
The presentation of a primary APS progressing into SLE is infrequent and is not commonly reported. Therefore, close follow up should be done for patients with primary APS, especially those who present with hematologic manifestations such as thrombocytopenia, neutropenia, and hemolytic anemia. The inability to resolve the pulmonary embolus through anticoagulation may lead to a rare complication of thromboembolism, which is CTEPH, which would require surgical intervention. Though the standard of treatment for thromboembolism in patients with APS requires warfarin, such cannot be used in this patient due to a lack of facilities for monitoring therapeutic levels. The introduction of DOACs has elucidated some alternative management in anticoagulation in patients with APS. However, further studies should be made to clearly define the benefits and risks of DOACs in APS related thromboembolism.