Human Immunodeficiency Virus (HIV) and Acquired immunodeficiency syndrome (AIDS) is a major cause of morbidity and mortality in Africa. Sub-Saharan Africa accounts for over 27 million (75%) of the global burden of HIV. Viral co-infections are also becoming common especially Hepatitis B virus (HBV) and hepatitis C virus (HCV) viruses due to shared risk factors and mode of transmission. HIV and Hepatitis co-infections affects disease progression and complicates management. Genotypes of HBV are associated with variations in clinical outcomes, natural course of infection, disease progression and treatment responses. For HIV/HBV co-infected patients, there is a high risk of developing severe liver disease.
Objective: The study aim was to determine HBV genotypes among HIV infected patients in multiple study sites in Kenya.
Methods: Study participants were recruited from patients on comprehensive care in selected health facilities. Blood samples collected were shipped to HIV Research Division laboratories, Centre for Virus CVR), at Kenya Medical Research Institute [KEMRI] where samples processing, genotyping and analysis was carried out.
Results: This is the second time that HBV genotype C is being reported in Kenya. While genotype A is the predominant genotype in this study, we also identified genotypes D as circulating within the study population Total sequenced samples were 71, genotype distribution included 67.6% genotype A [n = 48], 19.7% genotype C [n = 14] and 12.7% genotype D [n = 9].
Conclusion: The study observed diverse HBV genotypes among HIV co-infected individuals, which necessitates continuous surveillance and monitoring for better management of this population.
HBV, Genotypes, Co-infections, HIV, Testing, Sequenced, DNA
HBV: Hepatitis B Virus; HCV: Hepatitis C Virus; HCC: Hepatocellular Carcinoma; DNA: Deoxyribonucleic Acid; HIV: Human Immunodeficiency Virus; CCC: Comprehensive Care Clinic
Hepatitis B Virus, the smallest human DNA virus with a genome of 3200 bp is one of the most common viral infections. Based on its global distribution, HBV has gained its place as a disease of public health importance. The global prevalence of HBV infections has been reported to range from 2% to 20% [1]. Besides the high prevalence, the other important characteristics of HBV infections include; persistent infections, a wide spectrum of clinical manifestations including inactive carrier state, chronic hepatitis, liver cirrhosis and hepatocellular carcinoma (HCC). Consequently, about 15%-40% of HBV carriers have a lifetime risk to develop cirrhosis, liver failure, or HCC [1,2]. When people with chronic HBV mono-infection are compared to HIV/HBV co-infected individuals, the later are known to have a rapid advancement to liver disease and end-stage liver disease. Chronic hepatitis B infection has been observed in approximately 8% to 10% of HIV infected individuals worldwide. The prevalence in HIV/HBV coinfection differs by region with Sub-Saharan Africa coming second globally at 10.0% while Western Pacific region rates the highest at 11.4% [3].
Based on divergence in nucleotide sequence of about 8% and 4%, HBV has been divided into ten genotypes [A-J] and over forty sub-genotypes [4,5]. Significance of the genotypes is their variation in clinical outcome where the natural course of infection, disease progression and treatment responses could be different [6].
Genotypes B, C and I have been implicated in frequent vertical transmission; genotypes A, D and G is higher in sexual transmission and injecting drug use [4]. Genotypes A and C have higher chronification rate than genotypes B and D [7]. A faster progression to liver cirrhosis and HCC is associated with genotypes C, D and F [8]. Consequently, monitoring of circulating genotypes is important. Detection and identification of new genotypes in areas where there is limited information is necessary in order to improve hepatitis management strategies.
The global picture of HBV genotypes point towards a virus that selectively circulates more predominantly in some regions or countries. By distribution, genotypes A, D and E are the main HBV genotypes in Eastern Africa while A and E are mostly found in central Africa. Genotypes E and D are commonly found in North Africa, genotype A in Southern Africa and E in West Africa. In Asia, genotypes C, B and D are predominant while genotypes H and G are reported in Europe and America [4,9]. In East Africa, distribution of HBV genotypes is not well known, since only few studies have been conducted. Research to identify circulating genotypes, their distribution pattern and possible impact of these genotypes is a crucial gap in management and control of hepatitis. This study aimed at identifying genotypes of HBV in different regions in Kenya.
Study participants were recruited using simple random sampling criteria from HIV infected patients attending comprehensive care clinics (CCCs) in selected regions in Kenya. These selected regions broadly represented the Coastal, Central, Rift valley and Western parts of Kenya. Participants were taken through the consenting process and those who accepted to participate were recruited.
Blood samples were collected from the study participants. The samples were then separated by centrifugation and aliquots shipped to HIV Research Division laboratories, CVR at Kenya Medical Research Institute [KEMRI] for downstream processing and analysis.
All samples were tested for HBV by Rapid test using Determine HBsAg (Alere, USA) and Hepanostika ® HBsAg Ultra sensitive ELISA test for Hepatitis B infection (bioMérieux) following manufacturer’s instructions. For positive samples, HBV DNA extraction was carried out using Qiagen RNA/DNA extraction kit according to the manufacturers’ instructions (QIAamp ® Viral RNA/DNA mini kit Qiagen RNA/DNA extraction protocol). Briefly, 560 µl of prepared Buffer AVL containing carrier Nucleic acid (NA) was pipetted into a 1.5 ml micro centrifuge tube. This was followed by addition of 140 µl of plasma. This mixture was pulse-vortexed and incubated at room temperature for 10 min. 560 µl of ethanol (96-100%) was added to the sample, and mixed by pulse-vortexing. 630 µl of the extract was added to a QIAamp Mini spin column (in a 2 ml collection tube) and centrifuged at 6000 × g (8000 rpm) for 1 min and the supernatant discarded. The QIAamp spin column was placed into a clean 2 ml collection tube. 500 µl of wash buffer (Buffer AW1) was added and centrifuged at 6000 × g (8000 rpm) for 1 min. The supernatant was discarded and 500 µl of wash buffer (Buffer AW2) added and centrifuged at full speed (20,000 × g; 14,000 rpm) for 3 min. The QIAamp Mini spin column was placed in a new 2 ml collection tube. 60 µl of elution buffer (Buffer AVE) equilibrated to room temperature was added and incubated at room temperature for 1 min. This was then centrifuged at 6000 × g (8000 rpm) for 1 min to elute the viral NA. The DNA obtained was kept at - 80 °C. Polymerase chain reaction (PCR), primers and downstream application procedures were carried out as previously described [10]. Briefly, the extracted viral DNA was subjected to nested PCR with an AmpliTaq Gold PCR kit (Applied Biosystems, Foster City, Calif., USA). Amplification targeted preS1 gene of HBV. PCR products were subjected to gel electrophoresis. Amplification was confirmed by visualization under UV illuminator after staining in ethidium bromide. The PCR amplicons obtained were directly sequenced in an automated DNA sequence (ABI3100 Applied Biosystems, Foster City, CA) with BigDye Terminator version 3.0 Cycle Sequencing Reaction Kits (Applied Biosystems, Foster City, CA). Obtained sequences were then aligned using clustal W with reference sequences from Hepatitis B database to determine their genotypes. This was followed by construction of a phylogenetic tree by neighbor-joining method as previously described [10]. Resulting tree was visualized using tree view application.
This study was approved and cleared by Kenya Medical Research Institute, Scientific and Ethical Review Unit (SERU) (protocol number KEMRI/SERU/CVR/015/ 3581 ). Approvals were also sought from the respective Counties and heads of health facilities where the research was conducted. Study patients provided consent before participating in the study. Confidentiality of participants’ data was maintained.
Participants attending HIV Comprehensive care clinics from selected regions in Kenya were recruited as previously described [11]. The selected regions broadly represented the Coast, Central, Rift valley and Western parts of Kenya. Out of 1,000 samples screened, 150 tested positive for HBsAg, and 71 of these samples were successfully amplified by PCR and thereafter they were sequenced. The 150 HBV positive samples indicate dual infection.
Genotypes of hepatitis B (HBV) detected were A, C and D where Genotype A was predominant. A total of 71 samples were sequenced and the genotype distribution included 67.6% genotype A [n = 48], 19.7% genotype C [n = 14] and 12.7% genotype D [n = 9].
A rooted Phylogenetic tree illustrating the evolutionary relationships of hepatitis B genotypes found in the study in relation to reference sequence from hepatitis B virus database is illustrated in Appendix 1- Figure 1 below.
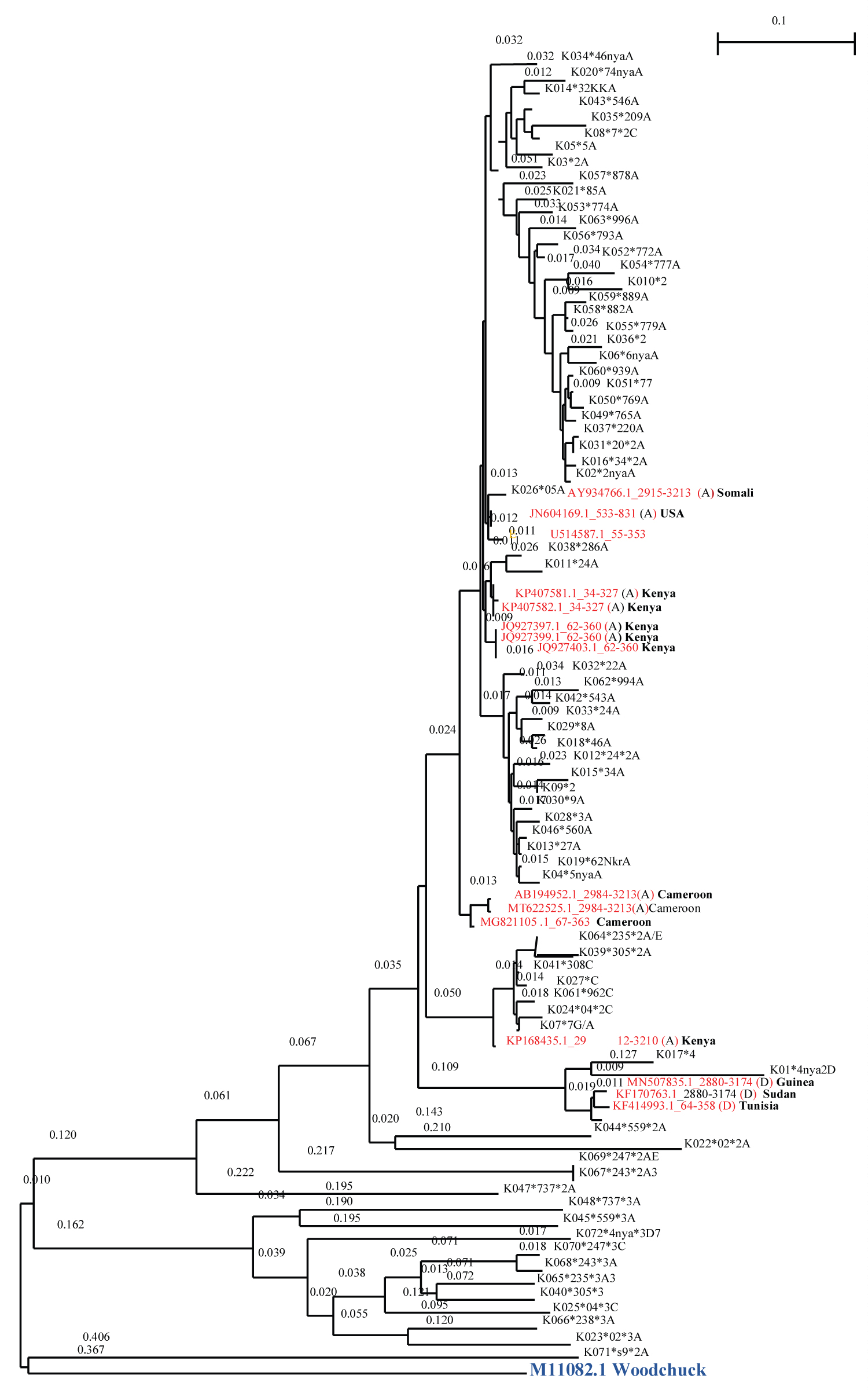 Figure 1: Phylogenetic tree of HBV.
View Figure 1
Figure 1: Phylogenetic tree of HBV.
View Figure 1
Phylogenic tree of HBV genotypes based on PreS1 region. The internal branches defining the genotype were supported by 1000 bootstraps. Reference sequences are labelled in red using their gene bank accession number and the country in which the sequences originated.
The study samples are labeled with K (for Kenya) followed by the patients code, then an asterisk (*) followed by health facility number or facility initials depending on sample collection site.
In this study, samples were obtained and screened for Hepatitis B virus from a population of HIV infected CCC attendees. The study describes HBV genetic diversity among HIV infected participants. The infection with HBV was 15%, which is within the reported prevalence of HBV in high endemic areas. The disparity in numbers of positives in serology {150} and PCR {71} could be due to low viral load arising from antiretroviral therapy.
Sequence analysis of genotypes identified HBV genotypes A, C and D to be circulating within the study population. Most studies on HIV/HBV coinfections and even HBV mono-infection have reported similar results [10,12-16]. Presence of previously undetected HBV genotype C could mean the genotype is newly introduced in the country or has continuously remained undetected in previous studies. Genotypes contribute differently to chronicity of infections and their long-term consequences. For example, variation of genotypes within a locality is also likely to contribute to intergenotypic recombination [1,17]. Recombination complicates disease outcomes since they are likely to escape immune surveillance efficiency in the body leading to increased transmission.
Monitoring of genotypes within a country is likely to help predict the severity of liver disease hence contributing long-term planning for various interventions. Some of the genotypes identified in this study including genotypes A, B, C and D have been shown to have high mutation rates than the rest of genotypes [18,19]. This is crucial for treatment guidelines and identification of suitable drug regiments.
Out of all the genotypes identified in this study, HBV genotype A was more predominant in all the study regions. This indicates that this genotype has a countrywide distribution. The wide distribution of this genotype is an important finding since it will provide guidelines to county health programs in prioritizing hepatitis management based on HBV genotypes endemic in their regions.
Genotype A in the phylogenetic tree clustered with references from Somalia, Cameroon, USA and Kenya. The clustering with Somalia could be because it is a neighboring country thus likely to be sharing the same genotypes due to the interactions between the two Countries. Kenyans also interact with the USA and Cameroon, which may explain the similarity in hepatitis genotypes. There was no particular pattern observed on the distribution of genotypes D and C in the regions under study. Since the first ever publication on genotypes of HBV in Kenya by Mwangi and team [10], genotype A has predominantly been reported in subsequent studies and in various sites [20-23]. This genotype is also the most predominant in East Africa region [4] and a major contributor to circulating genotypes of HBV globally. Globally, genotype A contributes to approximately 16.9% of all genotypes, coming in third after Genotype C (26%) and Genotype D (22%) in that order [4]. In the current study, percentage distribution of genotypes was 67.6% A, 19.7% C and 12.7% D respectively. In three of the previous studies in Kenya, genotype A, contributed to 90%, 88% and 69% of the total genotypes identified [10,15,20]. Many studies including this one have reported genotype A to be high in both HBV mono-infected individuals and HIV/HBV infected individuals. This indicates that this genotype continues to circulate in the population regardless of the locality or geographical region in the country. Genotype A is mostly transmitted sexually [18,24] and it has the ability to advance to persistent infections [25,26]. Unprotected sex is a major risk factor for hepatitis B (HBV) transmission. Up to 40% transmission rate occur from infected to non- immune partners of HBV infection. Moreover, biomarkers of HBV have been positively correlated with a number of lifetime sexual partners [27,28]. These risks may perpetuate continued circulation of Genotype A in the region. Consequently, increased mortality and morbidity could ensue [25,26]. Therefore, the future of genotyping for HBV in Kenya should explore the drivers and efficiency of transmission of this genotype.
Genotype D has been reported previously in Kenya [10,16,20]. In this study genotype D clustered with references from Guinea, Sudan and Tunisia. This could be because of human interactions between Kenya and these Countries. The worldwide distribution of HBV genotypes ranks genotype D as the second most distributed (22%), making it one of the most important genotypes in epidemiology of HBV infections. Detection of this genotype in this study is therefore an important finding since it is likely to contribute to significant proportions of HBV genotypes in future. Genotype D and A are mostly horizontally transmitted and have low hepatitis B e antigen (HBeAg) positivity. Status of HBeAg is important since it correlates with active infection and has implication on clinical outcomes including chronicity and control of HBV infection [1]. Identification of these genotypes in a population can guide on laboratory tests workup for management of HBV in a locality. Since genotypes A and D are mostly horizontally transmitted, tailored prevention and control interventions can be targeted within high risk populations.
This is the second study in Kenya to report on presence of HBV genotype C in the population after Kasera, et al. [16]. Genotype C accounts for majority of HBV genotypes globally (26%) and is known to have a lower response rate to treatment and high levels of disease progression [29]. This genotype is more oncogenic than genotypes A, B, and D [18]. In the current study, genotype C was second highest in distribution among the patients sampled. This could mean that this genotype is rapidly spreading in the population undetected or could be a new entry. Imperatively, detection of this genotype in the Kenyan population has public health importance especially in control and management of hepatitis. Nevertheless, its entry in the country may lead to rapid transmission and complication in management. Further studies on the genotype are important in order to elucidate its occurrence and monitor its circulation pathways.
Other genotypes of HBV including F to I, have not been reported in Kenya to date. This however, does not indicate their absence in the population. This is because global travel, tourism and cross border, events are common. Their significance however remains important as they may be existing; undetected, in very low percentage or have not yet been introduced in the country. HBV genotype B has so far been detected in one study in Kenya [16]. Distribution of genotypes E, B and F to I indicate they are responsible for 17.6%, 14% and 2% [4] of global chronic HBV infections respectively. Genotype E that was first identified in Kenya by Mwangi, et al. 2009, and subsequently in other studies was not detected in the current study. Globally it is an important genotype since it’s responsible for approximately 18% of chronic HBV infections. Genotype E is predominant in West Africa, where its origin is likely [30].
Findings of this study indicate HBV genotype A is still the predominant genotype in Kenya. Additional HBV genotype D and C were identified as well. This study is the second to identify presence of genotype C in the Kenyan population. Since this study covered many sites in the country, it is most likely that the findings represent local distribution of genotypes in the population. Surveillance of genotypes in Kenya is required in order to identify introduction of new genotypes and monitor circulation of the existing ones. A nationwide study is recommended in order to determine variants and extent of hepatitis B genotypes across the Country.
Surveillance of HBV genotypes is recommended especially where trends are changing and prevalence is high.
The authors declare no conflict of interest.
Authors wish to knowledge all participants in the study. We acknowledge colleagues in the ministry of health facilities where samples were collected for their efforts and support. Thanks to the management of comprehensive care clinics for supporting and providing access to their facilities and clients. Sincere thanks to Director, KEMRI and management and the government of Kenya for the small grants programme that makes such studies a success.
Internal Research Grants [IRG] of Kenya Medical research Institute provided funding support for this study.
Authors contributed equally to this publication.
Kenya Government funding (Internal Research Grants [IRG] of Kenya Medical research Institute).