Sickle cell disease is characterized by a very heterogeneous clinical course among patients with the same mutations for sickle cell hemoglobin (HbS). Sickle cell anemia (SCA) is a hereditary hemoglobinopathy caused by the homozygosity of a point mutation in the beta-globin gene, which leads to the substitution of glutamic acid for valine in the sixth position. Thus, this mutation results in an individual with the abnormal Hb phenotype and different physicochemical properties being known as HbS. The molecular basis for the formation of HbS is known, however, the mutation alone (HBB, glu6val, rs334) is not enough to explain the heterogeneous phenotype found in patients with SCD, other factors such as HbF levels, genetic polymorphisms, drug use how hydroxyurea and environmental factors can help to better understand this hemoglobinopathy.
To study how these factors modulate sickle cell anemia expression, the present work aimed toinvestigate the association of molecular, hematological, and biochemical markers with the severity of sickle cell anemia.
182 male patients homozygous for the Hb S genotype were selected. Disease severity was calculated from the "Sickle Cell Disease Severity Calculator". In this study, hematological (Ferritin), biochemical (TBARS-Thiobarburic acid-reactive species) and molecular markers - SNP were used in the genes: [TNFα(-308 G > A), TGFβ (-509 C > T), eNOS (-786 T > C) and HFE - H63D (63 G > A), C282Y (282 G > A) and S65C (65 A > T)].
Some associations were stronger, for example with polymorphisms of TNF-α (-308 G > A) genes with the most predominant genotype being the homozygous mutant polymorphism [GG]. Patients with Moderate and severe severity had pain attacks with a frequency of two attacks per year on average. In contrast, the frequency observed in Mild patients was 3-5/year. The eNOS gene polymorphism (-786 T > C) showed a higher frequency of the [TT] polymorphism being associated with stroke. The use of hydroxyurea, as well as the levels of Ferritin, showed to be great agents associated with the degree of severity. Of the polymorphisms of the HFE gene studied, only the H63D (63 G > A) presented a mean associated with the degrees of severity, with the frequency of the W/H63D genotype being predominant in patients with a moderate degree.
This work showed that the degree of severity in SCA is associated with TNFα, Stroke, Pain Crises, TGFβ, TBARS, Number of infections/year, eNOS, Ferritin, and mutations in the HFE gene (H63D, C282Y, and S65C).
Genetic markers, Sickle cell anemia, Severity, TBARS
Hb: Hemoglobin; SCA: Sickle cell anemia; SCD: Sickle cell disease; Il: Interleukin; TNF-α: Tumor necrosisfactor α; HU: Hydroxyurea; TGF-β: Transforming growth factor-beta; eNOS: Endothelial nitric oxide synthase; TBARS: Thiobarbituric acid reactive substances; HFE: Homeostatic iron regulator
Hemoglobin (Hb) is a heterotetramer consisting of four alpha and beta subunits, each, composed of a protein fraction called globin and a prosthetic group, the heme [1,2]. Mutations in the globin gene cause changes in the synthesis of alpha and beta chains, producing structurally different proteins, characterized by variant hemoglobins [3]. One of these variants is Hb S. Its formation is caused by a point mutation in the beta-globin gene (HBB), which leads to the substitution of glutamic acid for valine in the sixth position in the chain. When the gene for Hb S occurs in homozygosis (Hb SS), individuals have a severe disease condition called sickle cell anemia (SCA). Thus, this mutation results in an individual with an abnormal Hb phenotype and different physicochemical properties [4].
The incidence of newborns, worldwide, with sickle cell disease (SCD), varies according to region and ethnicity. Among African Americans in the United States of America, 1 in 360 newborns is a carrier of the gene [5]. In Brazil, the incidence of SCD in newborns varies substantially between states, a fact explained by the ethnic heterogeneity of the Brazilian population [6]. In the state of Rio de Janeiro, the incidence among live births is 1 in 1,200, which is one of the states with the highest representation of children born with SCD [7] Therefore, neonatal screening for hemoglobinopathies with a focus on sickle cell anemia is essential for early diagnosis, which in turn provides a better understanding and management of the genetics and pathophysiology of the disease.
The pathophysiological basis and underlying inflammatory mechanisms, as well as evolutionary genetics, are still gaps that require further studies in patients with SCA. It is known that the striking complication of sickle cell anemia is pain, typically described as acute, chronic, neuropathic, due to comorbidity or a combination of two or more of them [8] and resulting from Vaso-occlusive processes [2,9]. However, other manifestations are observed such as functional asplenia, acute chest syndrome, pulmonary hypertension, stroke, cumulative damage to multiple organs, and low life expectancy [10-12]. Polymerization of Hb S is the main pathophysiological occurrence in SCA [13,14] in which there is a change in the physical properties of erythrocytes that causes them to acquire a sickle shape, resulting in hemolytic anemia and blockage of blood flow, particularly in small vessels, that can damage organs [4].
The inflammatory state is a set of adaptive responses of the organism's cells as a result of infection or tissue injury, to restore organ function and homeostasis [15-17]. In patients with SCA, the chronic inflammatory state is a consequence of several factors that articulate and feed each other, forming a permanent inflammatory cycle. The vascular endothelium is an important factor in the inflammatory process and vaso-occlusion in SCA [18,19] in which interactions between erythrocytes and endothelial cells, leukocytes, and platelets play a central role [4]. Thus, during the vaso-occlusion process, neutrophils may be essential in adhering to the endothelium, promoting the attachment of sickle erythrocytes to these cells, reducing blood flow, and promoting vaso-occlusion [20]. The expression of some integrins, with together fibrinogen and leukocytes, can stimulate effector functions such as phagocytosis, degranulation, and the production of pro-inflammatory cytokines/chemokines, such as interleukin 6 (IL-6), tumor necrosis factor α (TNF- α) and IL-1 [21].
A chemotherapy drug that significantly contributes to reducing the number of vaso-occlusive crises is hydroxyurea, which stops cell division [22-24] to compensate for low oxygen levels. In addition, it promotes a decrease in the number of circulating neutrophils and a reduction in the expression of erythrocyte adhesion molecules [25 ].
Sebastiani and contributors [26] proposed a predictive model (https://www.bu.edu/sicklecell/) to assess the severity of the Hb S genotype in American patients. This model was designed using Bayesian network modeling, in which the risk of death is estimated at five years as a disease severity score, which, in turn, can be used in studies involving clinical and laboratory variables and has shown high specificity and sensitivity. This tool was used in Brazilian populations [27] to classify patients with SCD according to the age range of the score established by the calculator.
Based on this context, this study aimed to verify the association of molecular, hematological, and biochemical markers with the severity, determined by the calculator in male patients with the Hb SS genotype.
From a universe of 1500 blood samples from patients with SCD, 500 were randomly chosen and classified according to the Severity calculator.
From the initial sample (500) we excluded those who were: 1) Smokers; 2) Alcohol drinkers; 3) Pregnant 4) Patients with stroke; 4) Patients with pain, 5) Patients with a hemolytic crisis. The time established for the exclusion criteria was four weeks before the date of sample collection.
In the selection of patients who used hydroxyurea, those who used the drug for more than 90 days with a mean of 41.1 months were selected, with the mean dose per patient being 26 mg/kg/day. To eliminate possible biases arising from different Hb genotypes, only homozygotes (Hb SS) were selected, performed on a total of 182 male patients, aged between 18 and 71 years and included. This manuscript is in According to the Clinical Protocols and Therapeutic Guidelines for Patients with SCD in Brazil [28].
To classify the disease severity scores in the selected patients, the "Sickle Cell Disease Severity Calculator" tool was used, which ranks patients according to their clinical phenotype (Mild, Moderate, Severe). The model considers the risk of death as a score, ranging from 0 (least severe) to 1 (most severe) [27].
Two electrophoretic procedures were used to identify the patient’s phenotype: In cellulose acetate at pH 8.6 and on agar at pH 6.2. Data of the Hb fraction quantification were obtained using high-performance liquid chromatography (HPLC) with the Trinity® Premier Resolution Analytical Columns equipment. Genotype was confirmed by PCR-RFLP [29].
Due to their importance in pathophysiology, the following parameters were selected for this study: Hematological (Ferritin), biochemical (TBARS-Thiobarburic acid-reactive species) and molecular markers - SNP in following genes: [TNFα (-308 G > A), TGFβ ( -509 C > T), eNOS (-786 T > C) and HFE - C282Y (282 G > A), H63D (63 G > A) and S65C (65 A > T)].
Hematological data and information onthe use of HU, clinical complications, blood transfusions, and exposure to environmental interferences were obtained by consulting the medical records and also the hematology service administration system (SASH) by the clinicians of the service, protecting the patient confidentiality.
Ferritin is a protein responsible for iron storage in the body and, for this biomarker, we considered reference values in healthy male patients of 23 to 336 ng/mL. For this, ferritin levels were considered normal (23 to 336 ng/mL), high (337 to 1000 ng/mL) and elevated ( < 1000 ng/mL). The values obtained in the biochemical tests were determined in fasting venous blood samples by routine methods.
The biochemical marker TBARS was estimated through the levels of lipid peroxidation evaluated in heparinized plasma according to the protocol proposed by Uchiyama and Mihara [30] and values up to 440 ng/mL were considered normal [31].
The detection of the -308G/A (rs1800629) mutation of the TNFα gene was performed by the PCR RFLP method followed by restriction analysis. The fragment obtained from PCR was 117 bp. The mutation in the -308G/A promoter region eliminates the restriction site of the NcoI Enzyme. Therefore, when the amplified fragment was submitted to enzymatic digestion, the normal allele generated two fragments: 97 bp and 20 bp, and the mutant allele only produced one fragment: 117bp [32].
The detection of the -509C/T (rs1800469) mutation of the TGFβ gene (-509 C > T) was performed by RFLP PCR followed by restriction analysis. The fragment obtained from PCR was 406 bp. The mutation in the -509C/T promoter region eliminates the restriction site of the Bsu36I Enzyme. Therefore, when the amplified fragment was submitted to enzymatic digestion, the normal allele generated two fragments: 223pb and 183pb, and the mutant allele only generated one fragment: 406pb [33].
The detection of the -786 T/C (rs2070744) mutation of the eNOS gene was performed by RFLP PCR followed by restriction analysis. The fragment obtained from PCR was 180 bp. The mutation in the promoter region -786T/C eliminates the restriction site of the MspI Enzyme, therefore, when the amplified fragment was submitted to enzymatic digestion, the normal allele generated two fragments: 138 bp and 42 bp, and the mutant allele three fragments: 92 bp, 46 bp and 42 bp [34].
The frequencies of the three variants of the HFE gene (C282Y, H63D, and S65C) were analyzed by the PCR RFLP method. To detect the C282Y mutation, the sequence of primers used were: L282 (sense): 5' GGG TAT TTC CTT CCT CCA ACC 3' and R282 (antisense): 5' CTC AGG CAC TCC TCT CAA CC 3'. The following primers were used to detect H63D and S65C mutations: L63 (sense): 5' ACA TGG TTA AGG CCT GTT GC 3' and R63 (antisense): 5' ATC CCC AGC CTT GTT AAC TG 3', according to protocols previously established [29].
IBM SPSS Statistics 20 and RStudio® software were used for the analysis. Data were presented in percentage, according to the frequency presented. We used the chi-square test of independence test between the level of severity and the dependent variables to verify whether or not there was an association. Due to the expected frequency of some variables to present low values, Fisher's exact test was also performed. The degree of association was assessed using Cramer's V test. Continuous variables were categorized for carrying out the tests. The results were arranged in descriptive tables and bar graphs. A significance level of 5% (p ≤ 0.05) was considered for all analyses.
Our results were compiled in Table 1.
Table 1: View Table 1
The degree of disease severity was significantly associated with all the variables presented in the table above (p > 0.05).
The degree of association between severity, clinical manifestation, and the investigated markers is shown in Figure 1.
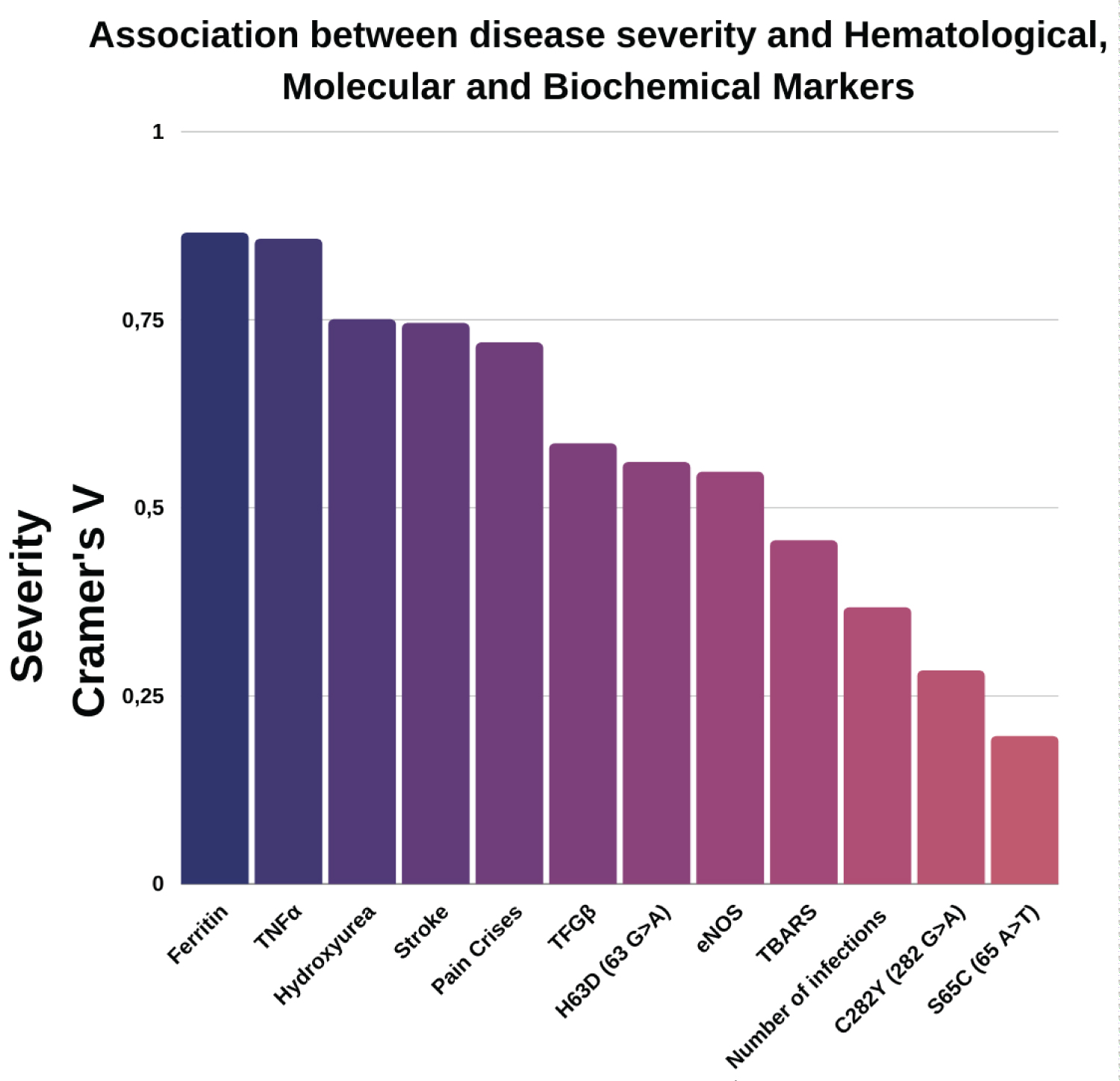 Figure 1: Association between severity score calculated by Cramer's V test and data from hematological, biochemical, and molecular markers. High values for Cramer's V2 indicate a stronger relationship between the variables, and lower values for V2 indicate a weak relationship. A value of 0 indicates that an association does not exist. A value of 1 indicates that there is a very strong association between the variables.
View Figure 1
Figure 1: Association between severity score calculated by Cramer's V test and data from hematological, biochemical, and molecular markers. High values for Cramer's V2 indicate a stronger relationship between the variables, and lower values for V2 indicate a weak relationship. A value of 0 indicates that an association does not exist. A value of 1 indicates that there is a very strong association between the variables.
View Figure 1
Some associations were stronger, for example, with the polymorphisms of the TNF-α genes [AA], [GA], and [GG], The most predominant genotype in the groups studied was the homozygous mutant polymorphism [GG] (Figure 2) in individuals who had a mild degree of severity.
 Figure 2: Demonstration of the frequency of genetic polymorphisms in which eNOS genes. C/C: Wild-type homozygote, C/T: Heterozygous, T/T: Homozygous mutant, TNFα: A/A: Wild-type homozygote, G/A: Heterozygous, G/G: Homozygous mutant, TGFβ: C/C: Homozygous wild, C/T : Heterozygous, T/T: Mutant heterozygote, H63D/H63D: Mutant homozygote, W/H63D: Heterozygous, W/W: Wild homozygote, W/S65C: heterozygous, W/W: Wild homozygote.
View Figure 2
Figure 2: Demonstration of the frequency of genetic polymorphisms in which eNOS genes. C/C: Wild-type homozygote, C/T: Heterozygous, T/T: Homozygous mutant, TNFα: A/A: Wild-type homozygote, G/A: Heterozygous, G/G: Homozygous mutant, TGFβ: C/C: Homozygous wild, C/T : Heterozygous, T/T: Mutant heterozygote, H63D/H63D: Mutant homozygote, W/H63D: Heterozygous, W/W: Wild homozygote, W/S65C: heterozygous, W/W: Wild homozygote.
View Figure 2
This high frequency of the [GG] genotype in mild patients was explained by recent work [35] that showed that the TNFα gene polymorphism (-308 G > A), specifically the [GG] genotype, is more frequent in healthy patients than in cancer patients, also an inflammatory condition, and that this genotype is linked to low production of TNF-α, so, the [GG] genotype would confer greater protection against the severity of the disease. The second most frequent polymorphism [GA] was observed in those patients who had a severe phenotype by the calculator. The heterozygous polymorphism [GA] is associated with the promotion of the activation of adhesion molecules in neutrophils and vascular endothelium [36] thus being able to be a risk factor for the occurrence of crises in SCA.
Our frequency of polymorphisms data corroboratesthose obtained in the Cavalcanti and contributors study [37], which from 25 patients with SCA in the northeast region of Brazil observed that SCA patients tend to have a higher production of TNF-α, a fact that can be explained by the chronic inflammatory processes even in patients in steady-state [38-40]; with the frequencies of polymorphisms [GG] > [GA] > [AA] > fact also observed in other studies [36,41]. These results are important as this high frequency/prevalence of the [GG] polymorphism can be used as a severe prognosis biomarker of serum TNF-α production.
Another fact with a strong association was pain crises, classified in frequencies of 0-2 and 3-5/year and greater than six/year. In our work, we observed that patients with a moderate severity had pain attacks with a frequency of 2 attacks per year on average. In contrast, the frequency observed in Mild patients was 3-5/year. This observation can be explained by the greater use of Hydroxyurea in patients with a moderate severity degree. The use of HU may be associated with peripheral pain sensitization [42,43] and other analgesic drugs [44]. It is known that one of the greatest difficulties faced by patients with sickle cell anemia is the pain (common in 70% of patients) [45,46] and these crises are be caused by vaso-occlusive processes, a complex phenomenon in which the interactions between erythrocytes and endothelial cells, leukocytes and platelets play a central role [4].
The analysis of the eNOS gene polymorphism (-786 T > C) showed a higher frequency of the polymorphism [TT] a wild-type genotype, followed by the heterozygote [CT], and finally, a lower frequency forthe variant allele [CC] in this patients. Similar data were reported by [47] who studied the impact of these polymorphic variants on nitric oxide activity in patients with sickle cell anemia. From these data, we can suggest that eNOS gene polymorphisms (-786 T > C) are important in the pathophysiology of SCA. There was an association with stroke (Figure 1) and these episodes were observed in patients with Moderate and Severe scores severity (Figure 3).
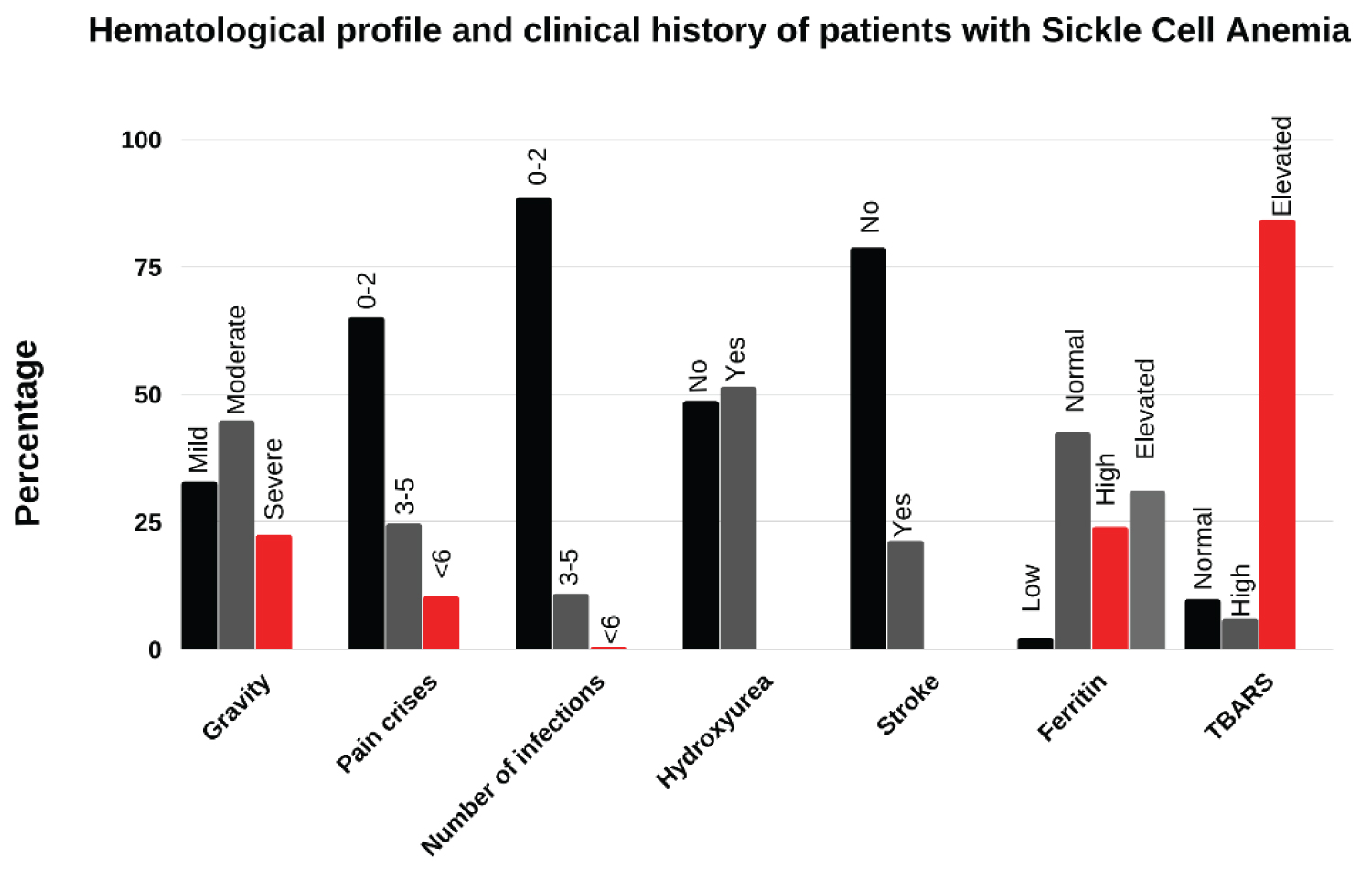 Figure 3: Percentage of the main components of the hematological profile (clinical complications, biochemical and hematological markers) of male patients with SCA as well as the percentage of patients with different degrees of severity according to the calculator. N = 182 patients observed.
View Figure 3
Figure 3: Percentage of the main components of the hematological profile (clinical complications, biochemical and hematological markers) of male patients with SCA as well as the percentage of patients with different degrees of severity according to the calculator. N = 182 patients observed.
View Figure 3
It is known that stroke is a major driver of mortality and morbidity in patients with sickle cell disease [48] a fact that could be explained by two hypotheses, the first is that the high rate of strokes would be due to the viscosity of sickle cell red cells, which would culminate in thrombosis and consequently ischemia [49]; however, this would not be enough to explain the origin of strokes in the great vessels, which are marked episodes in this disease. The second hypothesis concerns the connection of sickle cells to the vascular endothelium, where activation occurs by direct contact of sickle erythrocytes, heme, free Hb, and ROS induced by hypoxia/reperfusion [50]. This process influences the production of inflammatory mediators, such as IL-1β, IL-6, and tumor necrosis factor (TNF), which lead to a chronic inflammatory state that, in sickle cell disease, leads to a lower expression of anti-inflammatory proteins and increased expression of pro-inflammatory cytokines [51-54].
This stroke mortality rate could be explained by the high number of leukocytes, as well as the increase in the levels of adhesion molecules and inflammatory markers such as VCAM-1 [55,56]. Furthermore, it has been shown that TNF-α 308 [GA] is associated with an increased risk of stroke in patients with SCA [38] the same findings were observed in our results. Thus, if there was a high frequency of TNF-α 308 [GA] we would tend to have a higher percentage of stroke patients. A review carried out by the author [57] showed that there is controversy regarding the genetic, clinical, and laboratory factors associated with the development of stroke in individuals with SCD, a fact that could be explained by the lack of uniformity and standardization in the definition of events other than cerebrovascular diseases (ischemic stroke, TIA, hemorrhagic stroke, silent infarctions, vascular stenosis detected by ARM and moya-moya disease) making it difficult to interpret and compare the results of the studies.
The use of hydroxyurea, as well as the levels of Ferritin, were shown to be major agents associated with the degree of severity (Figure 1). Patients with moderate (48 patients) and severe (40 patients) scores of severity had the highest drug use. Normal ferritin levels (23 to 336 ng/mL) were predominant in patients with a mild score; high ferritin levels (337 to 1000 ng/mL) were predominant in severely ill patients, and very elevated levels ( < 1000 ng/mL) in moderately severe patients. It is known that the use of HU can improve the clinical conditions of patients with SCD by 98% [58-60] acting to increase fetal hemoglobin [61,62] by activating nitric oxide-dependent on soluble guanylyl cyclase [63]. It has been observed that the use of HU for two years can decrease high serum ferritin levels [64] in patients with sickle cell anemia. A similar study in beta-thalassemia patients reached the same results [58,64,65]. Interestingly, the high level of TBARS, prevalent in patients with moderate severity, could infer oxidative stress in the cellular environment, which results in the formation of highly reactive and unstable lipid hydroperoxides due to iron overload. Thus, although TBARS shows a median association with severity, it probably acts as an additional result of the increased value of iron and ROS in SCA.
Of the polymorphisms of the HFE gene studied, only the H63D showed a mean associated with degrees of severity, its frequency being predominant in patients with a moderate degree. The HFE gene is located on the short arm of chromosome 6 (6.p21.3) and collaborates in the regulation of iron metabolism, being a cofactor for several physiological and biological processes, and with a fundamental role in the manifestation of hereditary hemochromatosis (HH), an autosomal recessive disease characterized by excess iron in the blood [66]. The mutation to the C282Y polymorphism is a consequence of the transition from tyrosine to cysteine in codon 282 of exon 4. The H63D polymorphism results from the transversion of histidine by aspartic acid in the protein and the mutation occurs in exon 2 of codon 63. A mutation for the S65C polymorphism is responsible for the substitution of serine for a cysteine, due to the exchange in exon 2 [67]. These are the most frequent mutations for this gene in the Brazilian population [68-70].
The most frequent symptoms of iron overload are usually: fatigue, arthralgia/arthritis, abdominal pain, decreased libido, weight loss, hepatomegaly, splenomegaly, and arthropathy [71]. However, as iron deposition occurs gradually and cumulatively, the onset of symptoms occurs after the third decade of life, making early diagnosis of the disease difficult [72].
The HH diagnosis is confirmed by the presence of mutations H63D, C282Y, and S65C and is indicated for individuals with iron overload due to high serum ferritin concentrations, > 200 μg/L in women and > 300 μg/L in men. However, HH is mostly associated with homozygosity for the C282Y genotype, evidencing the clinical disease, and like the mutations for S65C and H63D has reduced penetrance, despite the mutation for H63D being more frequent when in compound heterozygosity with the C282Y gene (C282Y /H63D and C282Y/S65C) shows only moderate iron overload [73].
The HFE proteins resulting from the mutations alter cellular iron homeostasis and are related to several inflammatory and neuronal diseases. Especially in the brain, microglia and macrophages are responsible for maintaining iron homeostasis and inflammatory modulation associated with the pathogenic process of multiple diseases [74].
Thus, the genotypes of polymorphisms in the HFE gene fundamentally impact macrophage functions such as proliferation and survival and may increase the pro-inflammatory state [74] and consequently the release of mediators. This may be the mechanism by which HFE variants impact sickle cell disease status.
The genotypes in the HFE gene influence the severity of sickle cell anemia, especially the W/H63D genotype, more frequently in patients with moderate and severe degrees (Figure 1). Ferritin is high in inflammatory processes, such as those caused by hemochromatosis and hemolytic anemia [71]. Thus, the use of HU contributes to the improvement of severity in severe cases caused by genotypes in the HFE gene with high levels of ferritin, by reducing the number of leukocytes and the adhesion of red blood cells in SCA [59,75] preventing a worsening of iron absorption. In this way, the damage caused by oxidative stress would be reduced, with a consequent reduction in the level of TBARS. Since stress is a factor that may be involved in leukocyte recruitment, this is due to the release of iron, which initiates the hemoglobin oxidation pathway with damage to sickle cell erythrocytes [27] the combined results of polymorphisms and markers intensify vaso-occlusion and severity in SCA.
Our work showed that the degree of severity in SCA is associated with TNFα, Stroke, Pain Crises, TGFβ, TBARS, Number of infections, eNOS, Ferritin, and mutations in the HFE gene (H63D, C282Y, and S65C).
Furthermore, we did not find an association between the hydroxyurea use and the severity of the disease, with evidence of greater use of the hydroxyurea by patients with severe clinical conditions.The use of HU with an impact on the improvement of pain crises.
Some polymorphisms were more frequent in different degrees of severity, such as TNFα [GG] in patients with mild severity, and this genotype may provide greater protection against disease severity. In turn, the heterozygous polymorphism [GA] of TNFα may be a risk factor for the occurrence of crises in SCA; the stroke episodes were observed in patients with the moderate and severe scores; the high levels of TBARs displayed in patients with moderate severity; as well as the high levels of Ferritin and, finally, the polymorphisms in the HFE gene associated with moderate severity.
The association found between TBARS and severity evidences a probable relationship between the increased value of iron and ROS observed in patients with SCA.
Few studies and relative data regarding the association of these variables with disease severity were performed. Our data allow us to delineate a profile based on the severity of the disease with the most frequent clinical conditions in this segment of the population. The investigation of markers and their prognoses presented here allow health professionals to devise strategies for more effective support to patients, improving the quality of life of these individuals.
This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brasil (CAPES) - Finance Code 001.
The authors have no conflicts of interest to declare.
CAPES Higher Education Improvement Coordination
1. Jonathan de Oliveira Rios: Supported research planning and data collection in the field, laboratory and data analysis, as well as the writing of the article.
2. Thais Fernandes Ribeiro: Supported research planning and data collection in the field, as well as the writing of the article.
3. Gabriel Felipe Arantes Bertochi: Supported the statistical part and writing of the article.
4. Clarisse Lopes de Castro Lobo: Supported the writing and clinical aspects, of reading and writing of the article.
5. Claudia Regina Bonini Domingos: Supported research planning and field data collection, laboratory and data analysis, as well as supporting the writing of the article.