Thalassemia refers to a group of inherited diseases characterized by decreased or absent synthesis of normal globin chains. The direct consequence is an imbalance of the alpha and beta globin chain synthesis that results in anemia from ineffective erythropoiesis and hemolysis. The term thalassemia major refers to the severe form that is often associated with life-long transfusion dependent anemia. Hypogonadism is the most frequently reported endocrine complication, affecting 70-80% of thalassemia major patients. Hypogonadism is likely to be caused by hypertransfusion therapy resulting in iron deposits in the gonads, pituitary gland or both. However, hypogonadotropic hypogonadism resulting from iron deposition in the pituitary gonadotrope is more commonly found. Gonadal iron deposition in ovaries or testes occurs less frequently, as the majority of amenorrheic women can still ovulate after hormonal treatment. Despite recent advances in iron chelation therapy, excess iron overload in pituitary gonadotropic cells remains one of the major problems in thalassemic patients. Hypogonadism, mostly hypogonadotropic hypogonadism, is usually detected during puberty. Early diagnosis and treatment are crucial for normal pubertal development and to reduce the complications of hypogonadism. The risks and benefits of hormonal replacement therapy, especially regarding the thromboembolic event, remain a challenge for providers caring for thalassemic patients. We hereby present a case of 15-year-old unmarried girl with thalassemia major presenting with primary amenorrhea and poorly developed secondary sexual characteristics. A thorough history, clinical examination, laboratory and radiological investigations were conducted. These tests confirmed the diagnosis of hypogonadotropic hypogonadism. Patient was started on hormone replacement therapy. She is on regular follow-up and compliant with her treatment.
Thalassemia major, Hypertransfusion therapy, Iron overload, Hypogonadotropic hypogonadism, Primary amenorrhea, Hormonal replacement therapy
Thalassemia major is a hereditary disorder of hemoglobin synthesis that requires a regular blood transfusion. The combination of chelation therapy and blood transfusion in these patients has dramatically extended the life expectancy. However, the repetition of blood transfusions leads to the accumulation of iron in several organs such as liver, heart, and the endocrine glands, resulting in a high incidence of endocrine dysfunctions in children, adolescents, and young adults. In normal individuals, iron homeostasis is controlled mainly by iron absorption, not excretion. Lacking adequate excretory mechanisms, thalassemic patients receiving a blood transfusion (usually 1 mg of iron per 1 mL of blood) inevitably experience significant iron overload. As a consequence of iron overload in thalassemic patients, non-transferrin-bound iron (NTBI) is found excessively in the blood. NTBI is also a catalyst for the formation of reactive oxygen species, causing oxidative damage [1]. Studies of human anterior pituitary adenomas showed that gonadotropes require more iron as compared with other pituitary cell types [2]. Thus, these cells are most affected, resulting in declining synthesis of luteinizing hormone (LH) and follicle-stimulating hormone (FSH). Hypogonadotropic hypogonadism in thalassemia is related not only to iron toxicity on gonadotrope cells but also to adipose tissue and leptin. In addition to its effects on carbohydrate and fat metabolism and appetite, leptin also acts on the hypothalamic-pituitary-gonadal (HPG) axis. To our knowledge, several studies have been conducted on leptin levels in different age groups of thalassemic patients, and in all of them low leptin levels were observed [3]. The direct effect of iron, in particular that of NTBI, on the ovaries and testes is currently unknown. The ovarian reserve is preserved in the majority of female thalassemia patients, even in women with amenorrhea.
Primary amenorrhea resulting from hypogonadotropic hypogonadism occurs when the hypothalamus fails to secrete adequate amounts of gonadotropin releasing hormone (GnRH) or when a pituitary disorder is associated with inadequate production or release of pituitary gonadotropins [4]. Thalassemia major can produce hypopituitarism. This hypopituitarism leads to hypogonadotropic hypogonadism, an endocrinopathy occurring secondary to iron overload [5]. The iron overload is a consequence of frequent blood transfusion, which is the most important treatment modality for thalassemia major. Other possible causes of hypogonadism in beta thalassemia major include liver disorders, chronic hypoxia, diabetes mellitus and zinc deficiency [6]. The anterior pituitary is especially sensitive to increased iron concentration which disrupts the hormonal secretion leading to hypogonadism, short stature and acquired hypothyroidism [7]. We present a case of primary amenorrhea in a thalassemic patient to highlight that hypertransfusion and regular chelation therapy may have allowed improved survival in patients with thalassemia major but despite medical advances, growth failure and hypogonadism remains a significant clinical problem in these patients in adolescence.
A 16-year-old unmarried girl, a known case of thalassemia major at the age of 1, reported to the medicine outpatient department with primary amenorrhea and poor development of secondary sexual characters. She gave history of receiving monthly blood transfusions since she was 6 months of age and daily table to deferasirox as a chelating agent. She gave no history of weight loss, night sweats, headache, or visual disturbances. On examination, she was a thinly built, short statured girl with a height of 132 cm (52 inch). She had a hypoplastic nose with a thalassemic facies. The girl had mild pallor, but no thyromegaly, acanthosis nigricans, or galactorrhea. Her breast development was prepubertal (Tanner Stage I) and absent pubic and axillary hairs. The abdominal examination was within normal limits and undergone splenectomy 8 years back. Gynecological examination revealed the external genitalia to be prepubertal. Per speculum and vaginal examination was not carried out due to her unmarried status. However, uterus could be felt on per rectal examination.
Laboratory investigations including whole hematological and biochemical parameters were carried out which revealed hemoglobin of 7.5 gm/dl (normal 12-16 gm%), MCV-72.5 fL (normal 78-98 fL), MCH: 25 pg (normal 28-32 pg), RDW: 20% (normal 11-14%). Reticulocyte count was 13.2% (normal upto 2%). PBF showed anisochromia, anisocytosis including normocytes, microcytes with fair number of target cells, spherocytes, schistocytes and plenty of neucleated RBC's with normal white cells and platelets. Serum ferritin was raised 3138 ng/ml (N: 50-150 ng/ml). A detailed endocrinological profile was also conducted which was suggestive of hypogonadotropic hypogonadism as evidenced by FSH 1.66 mIU/ml (normal 3-8 mIU/ml) LH 1.01 mIU/ml (normal 2-11 mIU/ml), Estradiol 10.0 pg/ml (normal 20.4 pg/ml). TSH was a bit raised 6.97 mIU/ml (normal 5.5-6.5 mlU/ml). Anti TPO was highly raised (116 u/l, normal less than 10 u/l) Ultrasonography of the pelvis revealed a infantile uterus (2.9 × 1.7 × 1.2 cm) with small sized both ovary.
X-ray chest showed cardiomegaly widened upper mediastinal shadow and rib in the rib appearances (Figure 1). X-ray hand showed thinning of cortex and expansion of medullary cavity (Figure 2). MRI of the brain was suggestive of pituitary iron overload with a small sella and a reduced size of the pituitary gland.
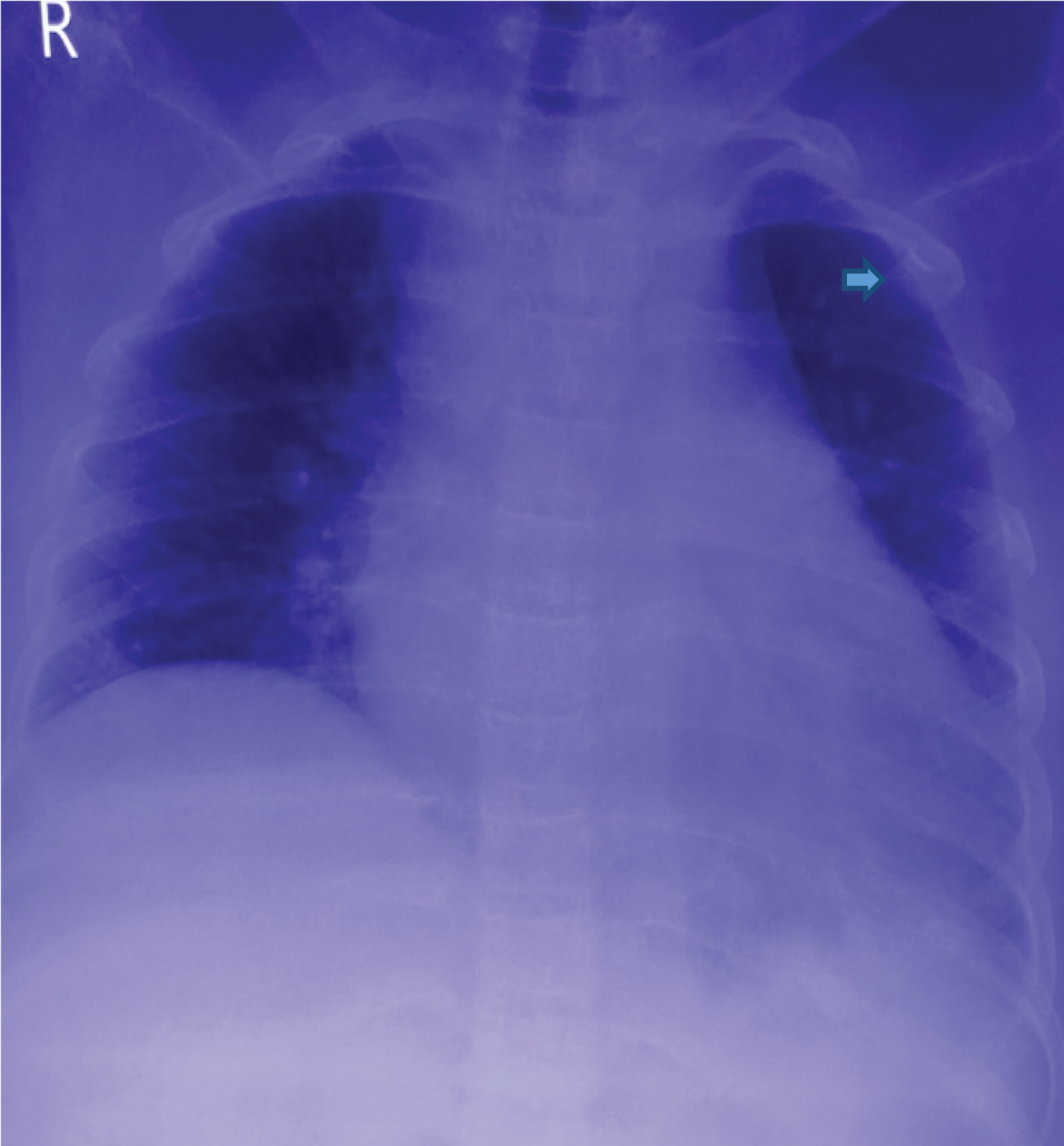 Figure 1: Chest X-ray PA view showing cardiomegaly, widended upper mediastinal and rib in rib appearance (arrow head).
View Figure 1
Figure 1: Chest X-ray PA view showing cardiomegaly, widended upper mediastinal and rib in rib appearance (arrow head).
View Figure 1
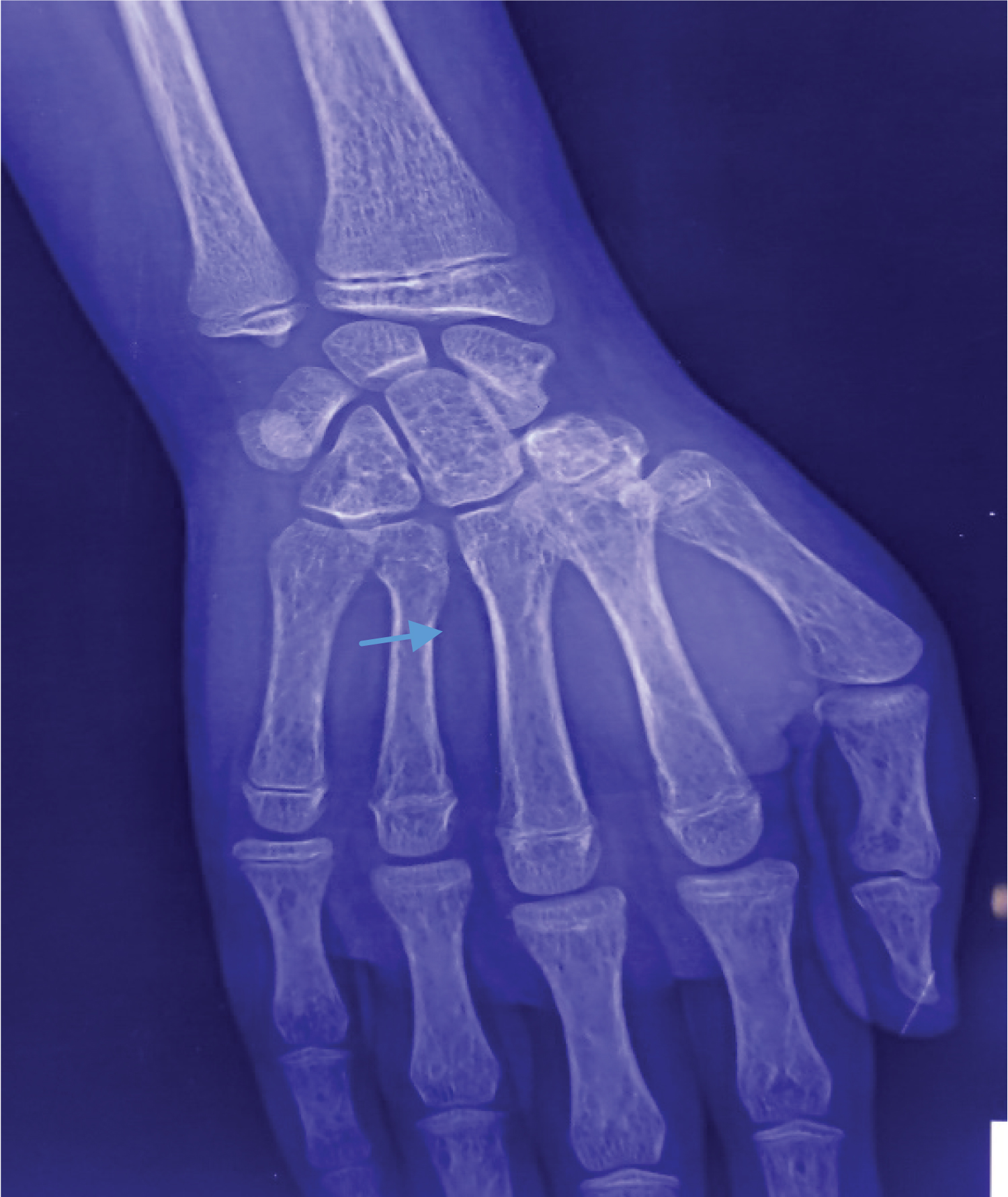 Figure 2 : X-ray of hand showing thinning of cortex and expansion of medullary caviy (arrow head).
View Figure 2
Figure 2 : X-ray of hand showing thinning of cortex and expansion of medullary caviy (arrow head).
View Figure 2
The clinical and endocrinological profile confirmed hypogonadotropic hypogonadism. Based on this diagnosis the patient was started on hormone replacement therapy to mature and maintain her secondary sexual characteristics. She was put on conjugated estrogens and cyclical progestins to prevent unopposed estrogen stimulation of the endometrium. She was also given low dose thyroxine supplement. Patient is on regular follow-up and compliant with her treatment.
The thalassemia's are a heterogeneous group of genetic disorders in which the production of one or more globin chains of hemoglobin is suppressed. Depending on the defective chain, several types of thalassemia's have been described like α and β thalassemia. Before the introduction of regular blood transfusion, thalassemia patients died within the first few years. Frequent transfusion and chelation therapy is the most important treatment modality which has considerably prolonged. The survival of thalassemic patients [8]. However, as a result of hypertransfusion therapy and prolonged longevity, iron tissue toxicity has become more common and contributes significantly too many structural, metabolic and endocrine consequences in these patients like hypothyroidism, hypogonadism and diabetes mellitus [9]. Hypogonadism resulting from iron induced pituitary dysfunction is the most frequently reported endocrine abnormality in patients with β thalassemia major [10].
Iron deposition in the anterior pituitary gland can be demonstrated beginning in the first decade of life, but clinical manifestations are usually not evident until the onset of puberty. At the earlier stage, only a diminished gonadotropin reserve with intact gonadotropin pulse was observed [11]. There may be an asymptomatic phase of pituitary siderosis before hypogonadism occurs. Later, the gonadotropin reserve significantly diminishes, with markedly reduced spontaneous pulsatile gonadotropin activity which may lead to irreversible damage of the HPG axis.
Hematologic phenotypes were significantly associated with hypogonadotropic hypogonadism. A majority of patients (86.4%) with the β0/β0 hematologic phenotype developed hypogonadotropic hypogonadism, while there was only a 25% occurrence in the β0/β+ phenotype [12]. High serum ferritin levels of more than 2500 ng/mL during puberty was also found to be a risk factor for hypogonadism [13], with a 2.75 times greater likelihood of having hypogonadism compared with patients with serum ferritin levels less than 1000 ng/Ml [14]. Nevertheless, splenectomized individuals who had serum ferritin levels less than 2500 ng/mL also had high rates of endocrine disorders.
There are three main clinical presentations of the HPG axis derangement in thalassemia major, including delayed puberty, arrested puberty and hypogonadism. Delayed puberty is defined as the absence of any pubertal signs by 14 years in boys and 13 years in girls [15]. Arrested puberty is defined as the absence of further pubertal progression for more than 1 year after puberty has started. In patients with hypogonadism, spontaneous fertility is possible in well-chelated and well-transfused patients, but others with hypogonadotropic hypogonadism may need assisted reproductive techniques. Gonadal function is usually intact in patients with hypogonadotropic hypogonadism, indicating that ovulation or spermatogenesis can be induced by exogenous gonadotropin therapy. Hypothyroidism and diabetes mellitus also influence the outcome of fertility treatment, and specific treatments are needed.
Primary gonadal failure results from gonadal iron deposition. While secondary hypogonadism due to iron deposition on gonadotropic cells of the pituitary gland as revealed by poor response of LH and FSH to GnRH stimulation or a combination of both primary and secondary hypogonadism. Some studies have reported the incidence rate of failure of onset of puberty reaching 50% and may approach even 100%. Leptin is a polypeptide hormone produced by adipose cells that acts as a permissive signal to initiate puberty. Impaired in Leptin synthesis has been stated caused by iron toxicity on adipose tissue which delays a sexual maturation. Manifestations of low gonadotropin levels are a delayed onset of menarche, oligomenorrhea, secondary amenorrhea, reduced testicular size and breast size. These manifestations usually occur in significantly elevated serum iron and ferritin levels [16].
Another common endocrine dysfunction was short stature. Around 20%-30% patients with thalassemia major were reported to have growth hormone (GH) deficiency. The remaining 70%-80% had provocative tests such as clonidine or glucagon stimulation tests have revealed a lower peak of GH levels compared to those found in patients with short constitutional stature. The yearly growth velocity is either decreased or completely absent in thalassemia patients. Factors that potentially caused growth failure include iron overload, Deferoxamine toxicity, free radical toxicity, anemia, zinc deficiency, delayed puberty, liver cirrhosis, primary hypothyroidism and defect in Growth Hormone- Insulin-like Growth Factor-1 (GH-IGF-1) axis [16]. Low serum insulin-like growth factor-1 with normal growth hormone reserve in short thalassemia patients indicates that a state of relative growth hormone resistance exists [17]. Skeletal maturation, on the other hand, occurs under the primary control of hormones from the adrenal, thyroid and sex glands [18]. This patient showed normal IGF-1 levels, suggesting that the short stature was not secondary to decreased IGF-I by the liver.
Thyroid dysfunction was a complication in 3-27% of thalassemia patients, but its severity is related to the degree of iron overload. Autoimmune has no role in the pathogenesis of hypothyroid in thalassemia patients. Most of thalassemia patients have subclinical compensated hypothyroidism with high TSH but normal T4 and T3 levels as in our patient. Only 5% of thalassemia patient develop overt clinical hypothyroidism that requires treatment. The pathogenesis is remained unclear but thought to relate to lipid peroxidation, oxidative stress, and free radical release. Thyroid antibodies are negative, the thyroid glands are impalpable, and clinical features of disease are not present [16,19].
HPG axis dysfunction can manifest as low estradiol or testosterone with low to normal serum LH and FSH as commonly seen in hypogonadotropic hypogonadism. Low estradiol and testosterone accompanied with increased serum LH and FSH indicates primary gonadal failure. In women, Anti-Mullerian hormone (AMH) AMH corresponds well with antral follicle count and can be used to accurately assess the ovarian reserve, independent of gonadotropin levels. AMH levels in thalassemia patients are overall normal, signifying that ovarian reserve is preserved and possibly serving as an important biomarker for reproduction [20]. The history and clinical findings are substantiated by the laboratory investigations which was present in this patient. The circulating gonadotropins are inappropriately low and the levels of estradiol are extremely below the normal values.
The role of pituitary magnetic resonance imaging (MRI) in thalassemic patients has been studied in recent years. Hypogonadotropic hypogonadism is often hard to recognize before puberty because of the immaturity of the HPG axis [21]. Early detection of pituitary iron overload is important since hypogonadism is not fully reversible by iron chelation [22]. MRI has been used to predict asymptomatic iron deposition in the heart, liver, pancreas, and pituitary gland [23-25]. Decreased pituitary volume has been observed, which may be due to apoptosis of gonadotropic cells, failure of gonadotropic cells to grow properly, and also the possibility of suppressed leptin level. Patients with transfusion iron overload begin to develop pituitary iron deposition since during the first decade of life. However significant pituitary volume loss, using mean and standard deviation for a particular age, is not observed until the second to third decade of life. Thus, the critical time for MRI surveillance may be at 10-20 years of age when many patients rapidly accumulate pituitary iron [21]. For children under the age of 7 years, MRI data are lacking. It may show empty sella, decreased pituitary size.
Iron deposition in the anterior pituitary gland can decrease pituitary MRI signal intensity significantly in the T2-weighted image [25]. MRI signal hypointensity is due to the paramagnetic effect of iron [26], and serves as a useful tool for early detection of pituitary iron overload [25].
Although liver iron concentration has been considered to be an excellent marker of total body iron load, no relationship has been found between liver and pituitary iron deposit using MRI [27,28]. The lack of correlation may be due to differences in transferrin receptor concentration, iron kinetics, and the degree of organ inflammation or fibrosis.
Sex steroid or pulsatile GnRH can be utilized to induce puberty if the HPG axis is functionally intact, especially at an early stage of hypogonadotropic hypogonadism. Later on, if the HPG axis is irreversibly damaged, sex steroid replacement therapy is the only option to induce puberty. Generally, it is advisable to initiate puberty with sex steroid replacement therapy by age 13 in women and age 14 in men [29]. Factors such as severity of iron overload, liver disease and growth hormone deficiency should be considered before pubertal induction [15].
According to the Thalassaemia International Federation recommendations [15], therapy in women may begin with oral ethinyl estradiol (2.5-5 μg daily) for 6 months, followed by hormonal reassessment. If spontaneous puberty does not occur within 6 months after the end of treatment, oral estrogen is reintroduced in gradually increasing doses (ethinyl estradiol from 5-10 μg daily) for another 12 months. If patients do not experience breakthrough uterine bleeding, low-dose estrogen-progesterone hormone replacement is the recommended treatment. Our patient being a young unmarried girl has been started on estrogens and cyclical progestin therapy. Timely recognition and prevention of the endocrine complications, by early and regular chelation therapy is mandatory for the improvement of the quality of life and favorable psychological outcome of thalassemic patients.
The risks and benefits of hormone replacement therapy should be discussed with patients. The high incidence of thromboembolic event in thalassemia, 1-29% from various studies [30], has led to the identification of a hypercoagulable state in thalassemic patients. The absence of the spleen can contribute to and increase the risk of thrombosis in many hematologic diseases. From the treatment of symptoms of the menopause clinical practice guideline of the Endocrine Society, randomized controlled trials (RCT) demonstrated that oral estrogen increases venous thromboembolism (VTE) risk in women aged 50 to 59 [31]. Observational studies and meta-analyses suggest that transdermal estrogen therapy does not increase VTE risk, even in women with thrombophilia. No RCTs were conducted in thalassemic patients; thus, hormonal replacement therapy in hypogonadal thalassemic patients should be used cautiously, preferably in transdermal form.
Early studies have shown little impact on the course of progressive endocrine dysfunction in patients with chelation therapy, Deferoxamine. Only aggressive chelation therapy with a combination of Deferoxamine and Deferipronehas achieved improved outcomes of endocrine complications. However, intensive chelation was normally in emergency and for a short period, as it could impact the therapy compliance negatively and drug-related adverse events [32].
High prevalence of endocrine disorders demonstrating that these abnormalities were related to iron overload. This patient has hypogonadotropic hypogonadism (resulting from iron deposition in the pituitary gonadotrope), short stature and subclinical hypothyroid, as his endocrine complication-related thalassemia. Endocrine evaluation in these patients should be done regularly, and it is recommended to start chelating therapy during the first years of life in patients who have iron overload. Treatment for endocrine abnormalities needs to continue to optimize the quality of life of the patient and for normal pubertal development. Fertility is usually retrievable with treatment. However, the evaluation for fitness for pregnancy and preconceptional improvement of health are of paramount importance for optimum results.
None declared.