In recent years, the Polycomb group (PcG) of proteins has been revealed to be involved in the regulation of hematologic stem cell function and differentiation and have been broadly linked to hematologic malignancies. Polycomb proteins are histone modifiers that contain two multi-protein complexes: Polycomb Repressive Complex 1 and 2 (PRC1 and PRC2). As each PcG gene present multiple orthologs, distinct PRC1 and PRC2 sub-complexes exist in different differentiation stage and tissues. Aberrant expression or mutation of individual PcG gene is likely to result in alteration of the PRC composition that is crucial for its enzymatic activity and target selectivity. Considering the dramatically increasing data on the regulation and functions of polycomb proteins, this review focuses on hematopoiesis and hematologic malignancies.
Epigenetic, Polycomb proteins, Hematopoiesis, Hematologic malignancies
The hematopoietic system is a well-established model for the homeostatic mechanism between self-renewal and differentiation. One Hematopoietic stem cell (HSC) could asymmetrically divide to a HSC destined for maintaining its line and to a multipotent progenitor, which could undergo expansion and generate large numbers of lineage-committed progenitors to consistently generate abundant numbers of new blood cells through the differentiation during our entire lifespan. In concept, one HSC is sufficient to generate the entire hematopoietic system.
Aberrant regulation of the differentiation of HSC could give rise to immortalized progenitors that lost proliferation control, leading to the development of hematologic malignancies, including leukemia, lymphoma, and multiple myeloma. Hematologic malignancies are originated from blood-forming tissue, such as the bone marrow, or in the cells of the immune system.
Although hematopoiesis and hematologic malignancies are regulated by multiple lineage-specific transcriptional factors [1], cancer genomic sequencing data from patients revealed that the frequency of genetic aberrations in hematologic cancer is much lower than most other malignancies, suggesting that epigenetic mechanisms are critical for hematologic malignancies [2,3]. Reciprocally, epigenetic modifiers that are aberrantly regulated in hematologic malignancies play a prominent role on hematopoiesis, including both self-renewal and differentiation of hematopoietic stem cells. One of the major epigenetic regulators is the class of polycomb-group (PcG) proteins that repress numerous genes involved in various biological process, such as cell differentiation, and cell cycle processes [4].
Polycomb group (PcG) proteins were first reported as an essential complex for controlling segmentation in drosophila [5]. In Drosophila, most of PcG target genes present specific cis-regulatory sequences named Polycomb-repressed elements (PREs) [6]. In mammals, PcG proteins incorporate two major functional complexes named polycomb repressive complex (PRC) 1 and 2 [3]. The canonical PRC1 complex consists of four core subunits sorted into four gene families including the CBX, PHC, PCGF and RING1. Each of these four core subunits presents multiple orthologs, which incorporate dynamic patterns of PRC1 complex depend on the differentiated status [7]. Through the ubiquitin E3 activity of its RING1 subunit, PRC1 mediates transcriptional repression by promoting the monoubiquitination of H2A at lysine 119 [8]. The PRC2 complex comprises three core subunits: suppressor of zeste 12 homolog (SUZ12), embryonic ectoderm development (EED) and one of two Enhancer of Zeste orthologs (EZH), EZH1 or EZH2 [9]. Through the histone methyltransferase activity of EZH2 or EZH1, PRC2 promotes the tri-methylation of H3 at lysine 27 (H3K27me3) generally associated with transcriptional repressive gene loci [10,11]. Intriguingly, both EZH1 and EZH2 have been revealed as transcriptional co-activators, suggesting that non-PRC2 function of EZHs may play an important role on RNA polymerase II elongation and drug-resistant of prostate cancer cells [12,13]. PRC2-mediated H3K27me3 is specifically recognized and bound by the chromo-domain of CBX subunit, thus recruiting PRC1 complex, which place PRC2 in the upstream of PRC1 (Figure 1A). RYBP is also known as RING1 And YY1-Binding Protein. It plays a role on the formation of non-canonical PRC1 complexes, which mediate H2A ubiquitination at polycomb target sites independent on PRC2 and H3K27me3 [14,15]. In addition, the RUNX1/CBFβ transcription factor complex mediates site-specific PRC1 chromatin recruitment also through PRC2-independent manner [16]. Intriguingly, it has been reported that PRC1-dependent H2AK119ub1 leads to recruitment of PRC2 and H3K27me3 to effectively initiate a polycomb domain [17], which place PRC1 into the upstream of PRC2 (Figure 1B). As various mechanisms have been found for recruitment of PRC1 and PRC2 in different cell types, the interdependence between PRC1 and PRC2 association at target gene loci is still an important issue to be addressed [18].
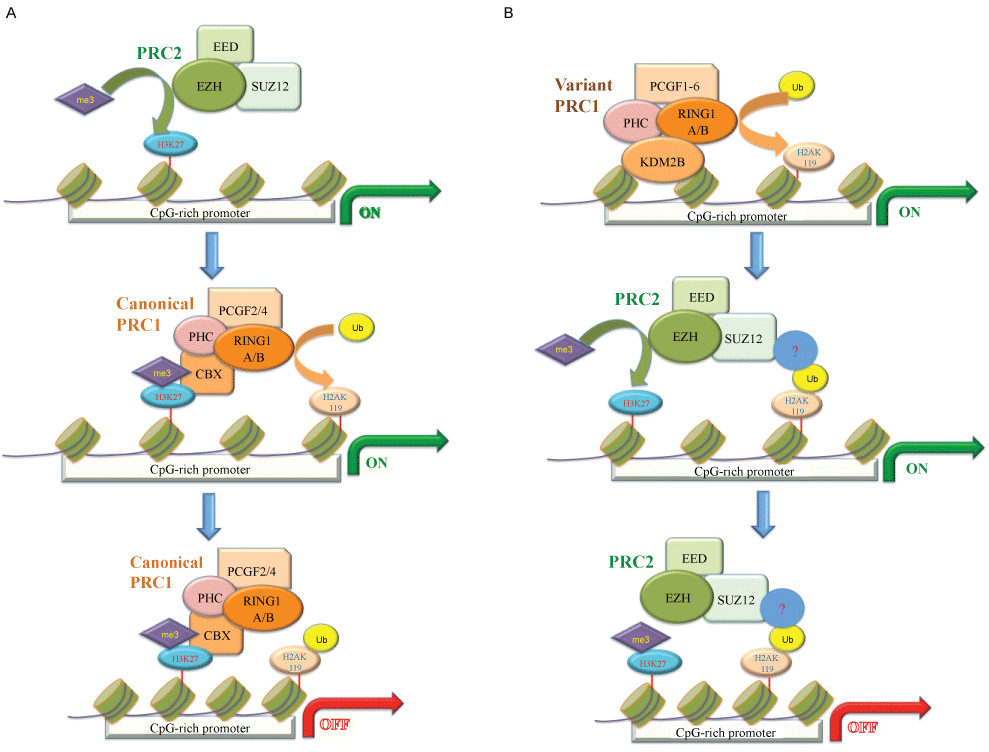 Figure 1: PRC1 and PRC2 can be recruited to chromatin in two different models. A) The "PRC2-dependent" model is based on the initial literature and implies that PRC2 mediates canonical PRC1 recruitment via H3K27me3 recognition. This scheme introduces the existence of PRC2-independent PRC1 sub-complexes that bind the same genomic loci independently of H3K27me3 and play a major contribution to sustain H2Aubq levels; B) The "PRC1-dependent" model shows that PRC2 can be directly recruited to chromatin by variant PRC1 sub-complexes, potentially by recognizing H2AK119 mono-ubiquitination. The subunit KDM2B in variant PRC1 directly binds to CpG-rich DNA.
View Figure 1
Figure 1: PRC1 and PRC2 can be recruited to chromatin in two different models. A) The "PRC2-dependent" model is based on the initial literature and implies that PRC2 mediates canonical PRC1 recruitment via H3K27me3 recognition. This scheme introduces the existence of PRC2-independent PRC1 sub-complexes that bind the same genomic loci independently of H3K27me3 and play a major contribution to sustain H2Aubq levels; B) The "PRC1-dependent" model shows that PRC2 can be directly recruited to chromatin by variant PRC1 sub-complexes, potentially by recognizing H2AK119 mono-ubiquitination. The subunit KDM2B in variant PRC1 directly binds to CpG-rich DNA.
View Figure 1
BMI-1 is crucial for the maintenance of self-renewal of hematopoietic stem cell (HSCs) [19]. The number of HSCs was markedly reduced in postnatal Bmi-1-/- mice and the self-renewal of adult HSCs was no detectable, indicating a cell autonomous defect in Bmi-1-/- mice [20]. Consistent to this phenotype, the expression of the genes associated with stem cell, cell survival and proliferation including p16Ink4a and p19Arf was altered in bone marrow cells of the Bmi-1-/- mice. Intriguingly, Bmi-1 directly targets the Cdkn2a locus, which encodes P16Ink4a/P19Arf [21]. Double knockout of Bmi-1 and Cdkn2a revealed a partial rescue of HSC function, suggests that the repression of cell cycle inhibitor P16Ink4a/P19Arf by Bmi-1 is critical in HSCs [21]. Cells derived from Bmi-1-/- mice also have impaired mitochondrial function resulting in a dramatically increase of reactive oxygen species and subsequent induce the DNA damage response mediated cell death, indicating that Bmi-1 may have a protective effect against oxidative stress that plays a crucial role in the self-renewal and survival capacity of HSCs [22]. In addition, loss of Bmi-1 lead to premature expression of B-lymphoid genes in progenitors accompanied by accelerated lymphoid lineage specification, indicating that Bmi-1 is a possible inhibitory factor of lymphoid lineage differentiation [23]. In contrast to Bmi-1, knockout of Mel-18, another PCGF gene family member, failed to cause apparent defect in the self-renewal of HSCs but increased proliferation of B-cells, suggesting that Mel-18 plays a role for in more differentiated cells [24]. Polycomb chromobox (CBX) family members show distinct expression patterns dependent on HSC-stage-specific, indicating various formations of PRC1 complexes present during HSC differentiation [25]. Transplanted CBX2-overexpressing HSCs in mice showed only B-cell reconstitution, suggesting that CBX2 is involved in lymphopoiesis, but not myelopoiesis [25]. CBX7 is highly expressed in the HSCs and plays an irreplaceable role in the self-renewal of HSCs by repressing the expression of progenitor-specific genes, suggesting that the PRC1 complex in HSCs preferentially contains CBX7 [25]. In contrast to CBX7, over expression of CBX2, CBX4 or CBX8 promotes the differentiation and exhaustion of HSCs, suggesting that other CBX proteins can compete with CBX7 to incorporate CBX2-, CBX4- or CBX8-containing PRC1 complexes that target the genes associated with the differentiation of HSCs [25]. On the other hand, the role of non-CBX containing PRC1 complexes in hematopoiesis is much less known. Recently, PCGF1 mediated transcriptional repression of Hox genes has been revealed to be required for the self-renewal in Runx1-/- HSCs, suggesting cooperation of transcriptional and epigenetic regulation is crucial for hematopoietic differentiation [26].
Comparing to PRC1, the role of PRC2 in hematopoiesis is mainly dependent on the EZHs proteins. Comparing to the ubiquitously expression of EZH2, EZH1 is a backup gene that is highly expressed in adult HSC but not in fetal HSC. Whereas EZH2-/- embryos died of anemia due to insufficient expansion of HSCs/progenitor cells and erythropoiesis, deletion of EZH2-/- in adult bone marrow had no alteration on hematopoiesis, suggesting that EZH1 complements EZH2 in the BM, but not in the fetal liver [27]. Deletion of Eed, a core subunit for the formation of both EZH1- and EZH2-containing PRC2 complexes, results in depletion of adult bone marrow HSCs while fetal liver HSCs are produced in normal numbers, suggesting that Eed present the EZH-independent function [28]. Although Eed-/- neonatal HSCs still expressed HSC signature genes, they were unable to differentiate into mature blood cells and were prone to cell death [28]. Deletion of Cdkn2a, whereas revealed partial rescue of HSC function in Bmi-1-/- mice, enhances hematopoietic stem/progenitor cell (HSPC) survival but fails to restore HSC functions in Eed-null mice [28]. These findings suggested that PRC2 plays a role during hematopoietic differentiation in a different manner with PRC1. Consistent with the role of PRC2 in transcriptional repression, Eed-/- HSCs present depression of PRC2 target genes largely associated with HSC self-renewal, differentiation and apoptosis, indicating that PRC2 suppresses genes of diverse pathways ensuring normal HSC functions [28]. Recently, it has been revealed that EZH1 and EZH2 undergo an expression switch mediated by GATA factors during blood cell development and loss of EZH2 expression results in repositioning of EZH1 chromatin occupancy and transcriptional activity (Figure 2), suggesting that the dynamic composition of PRC2 subunit leads to a switch from canonical repression to non-canonical activation during the differentiation from HSCs to progenitors [29]. More intriguingly, an EZH1-SUZ12 sub-complex lacking EED, named a non-canonical PRC2 complex, was identified to occupy active chromatin domains, and positively regulates gene expression [29]. In addition, EZH2 also plays a critical role in the early stage of B cell development and rearrangement of the immunoglobulin heavy chain gene (Igh) [30].
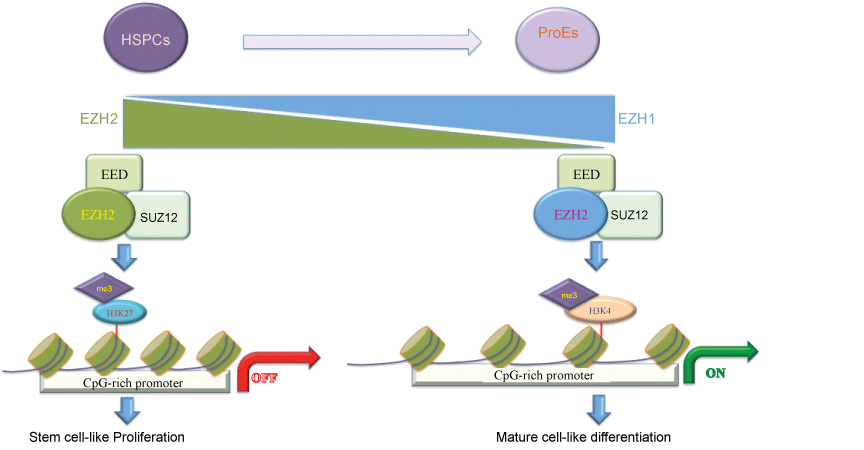 Figure 2: The dynamic PRC2 complexes switch during HSCs differentiation. This cartoon represents a switch from EZH2 to EZH1 expression during he differentiation of hematopoietic stem/progenitor cells (HSPCs) to highly enriched populations of erythroid progenitor cells (ProEs). In contrast to EZH2, EZH1 forms a non-canonical PRC2 complex and occupies active chromatin to enhance the expression of genes involved in differentiation.
View Figure 2
Figure 2: The dynamic PRC2 complexes switch during HSCs differentiation. This cartoon represents a switch from EZH2 to EZH1 expression during he differentiation of hematopoietic stem/progenitor cells (HSPCs) to highly enriched populations of erythroid progenitor cells (ProEs). In contrast to EZH2, EZH1 forms a non-canonical PRC2 complex and occupies active chromatin to enhance the expression of genes involved in differentiation.
View Figure 2
Bmi-1 has been found to be involved in leukemogenesis since it was recognized as a collaborator of c-Myc in the induction of B-cell lymphomas [31]. BMI-1 is commonly highly expressed in patients with myelodysplastic syndromes (MDS) [32], acute myeloid leukemia (AML) [33], chronic myeloid leukemia (CML) [34] and various types of lymphoma [35]. More intriguingly, BMI-1 expression is strongly correlated with disease progression and is associated with a poor prognosis in the patients with AML [33] and CML [36]. Studies in various leukemic mouse models suggested that Bmi-1 might be an important collaborating factor for leukemic transformation mediated by some fusion oncogenes, such as HoxA9-Meis1 [37], MLL-AF9 [38] and BCR-ABL [39,40]. In addition, Bmi-1 protects leukemia stem cells (LSCs) from senescence and apoptosis via repressing the expression of p16 and p19 expression [37,41]. Inhibition of BMI-1 expression mediated reactive oxygen species accumulation and apoptosis results in the reduction of proliferative capacity and stem/progenitor cell frequency in AML CD34 positive cells [19]. As BMI-1 could be efficiently inhibited by a small molecule in colon cancer cells in preclinical models [42], its required further studies to test which types of leukemia are susceptible to BMI-1-targeted therapies. Unexpectedly, deletion of Bmi-1 in Cdnkn2a-/- hematopoietic cells induced abnormal megakaryocytopoiesis accompanied by marked extra medullary hematopoiesis, which eventually resulted in lethal myelofibrosis, suggesting that Bmi-1 also present a tumor suppressor function [43]. As Bmi-1 may form different sub-complexes during hematopoietic cells differentiation.
Comparing to for BMI-1, less is known about other PRC1 components. It has been found that aberrant expression of PRC1 genes, such as MEL-18, RING1, HPH1, HPC1, in multiple types of lymphomas [44,45], suggesting that this abnormal formation of PRC1 contributes to the development of hematologic malignancies. Similar to Bmi-1, CBX8 has been shown to be essential in leukemogenesis induced by fusion oncogenes, such as MLL-AF9 [46]. CBX7 is often highly expressed in human follicular lymphomas (FLs) and its over expression in the mouse lymphoid compartment initiates T cell lymphoma and cooperates with c-Myc to induce highly aggressive B cell lymphomas [47]. Although it's remains unclear whether CBX4 is directly involved in leukemia development, it has been found that CBX4 expression is strongly correlated with the angiogenesis [48], metastasis [49] and prognostic [50] of hepatocellular cancer patients. Further studies found that CBX4 increased the transcriptional activity of hypoxia-inducible factor-1 (HIF-1) and enhancing the expression of HIF-1 target genes associated cancer metabolism, suggesting that CBX4 might play roles in other types of cancer [51]. RING1A, another PRC1 member, is commonly highly expressed in MDS and AML and is correlated with poor prognosis [32]. Additionally, SNPs in RING1A is correlated with non-Hodgkin lymphoma [52]. Enhanced expression of RING1B, a paralog of RING1A, has detected in multiple types of lymphomas [53].
Inactivating mutations of PRC2 components have been found in multiple types of hematopoietic malignancies. Patients with Early T-cell precursor acute lymphoblastic leukemia (ETP ALL) present high frequencies of mutations in PRC2 core components EZH2, EED and SUZ12 [54,55]. Inactivating somatic mutations of EZH2, EED and SUZ12 also occur frequently in patients with myelodysplastic disorders including myelodysplastic syndromes (MDSs) and MDS/myeloproliferative neoplasm (MPN) overlap disorders (MDS/MPN) [55-57]. In the patients with MDS/MPN, EZH2 mutations were frequently coincided with tet methylcytosine dioxygenase 2 (TET2) mutations [58]. Whereas deletion of EZH2 alone was enough to induce MDS/MPN-like diseases in mice, double depletion of EZH2 and Tet2 established more advanced myelodysplasia and dramatically accelerated the development of myelodysplastic disorders including both MDS and MDS/MPN [58]. Inactivating mutations of EZH2 are also frequently associated with RUNX1 mutations in MDS patients and loss of EZH2 significantly promote Runx1 mutant-induced MDS mice model [59]. EZH2-/- mice have been showed the phenotype that is associated with a thymocyte developmental block at the double negative stage and promote the development of T-lineage lymphoblastic leukemia [54]. On the other hand, hyperactive mutations of EZH2 have been detected in diffuse large B-cell lymphomas [60] and follicular lymphomas [61] in which EZH2 may be a potential therapeutic target [62,63]. Overexpression of EZH2 in transgenic mice has been showed to enhance myeloproliferation [64]. Knockdown of PRC2 subunits EED, SUZ12 or EZH1/EZH2 resulted in proliferation arrest and differentiation in different AML cell lines [65]. In MLL-rearranged leukemia, genetic mice models revealed that Eed is essential for leukemogenesis and leukemia maintenance, whereas EZH2 is dispensable for MLL-AF9 AML [66,67]. In summary, EZH2 plays both roles as pro-oncogene and tumor suppressor depends on different types of hematopoietic malignancies.
PcG proteins have become putative epigenetic regulators in both normal hematopoiesis and various hematological malignancies. The expression of PcG genes is highly regulated during the stage of hematopoietic cell differentiation. Aberrant expression or mutation of PcG genes is associated with different types of hematopoietic malignancies. Intriguingly, individual PcG genes display either tumor suppressor or oncogenic functions largely depending on the cell context. As each PcG gene present multiple orthologs, distinct PRC1 and PRC2 sub-complexes exist in different differentiation stage and tissues. Since distinct polycomb sub-complexes might target specific gene loci with different recruiting mechanisms, it is required more work to address the mechanism of the compositional switch and PRC recruitment. Overall, understanding the molecular mechanism of the genetic alterations and composition switch of polycomb proteins will not only provide important knowledge for hematopoiesis but also be beneficial for developing pharmacological method targeting of PRC compositions for the treatment of hematological malignancies.