The association hemolysis and venous thrombosis remains unknown to clinicians, despite our advances in comrehension of pathophysiological bases.
Haemolysis, which is observed in multiple diseases, can affect all three components of Virchow's triad. It is not surprising that there is a link between haemolytic disorders and thrombosis.
We will try to clarify the main pro-thrombotic mechanisms during hemolysis through 3 clinical observations of deep venous thrombosis in 3 main types of hemolytic pathologies, namely a case of paroxysmal nocturnal hemoglobinuria, thrombotic thrombocytopenic purpura and autoimmune anemia hemolytic.
The link between venous thrombosis and hemolysis seems multifactorial, involving mechanisms related to hemolysis and classic risk factors for Venous Thromboembolic Diseases.
The interaction between these risk factors deserves careful study given its implications in therapeutic decisions.
Hemolysis, Venous thrombosis, Thrombotic thrombocytopenic purpura, Autoimmune hemolytic anemia, Paroxysmal nocturnal hemoglobinuria
Hemolytic anemia is defined by the premature destruction of red blood cells (RBCs). Hemolytic anemia may be acute or chronic and life-threatening, and it should be considered in all patients with a normocytic or macrocytic anemia that is unexplained. Premature destruction of RBCs can be intravascular or extravascular in the monocyte-macrophage system of the spleen and liver; extravascular destruction is more common.
The mechanism of antibody-mediated hemolysis is via phagocytosis or complement-mediated destruction and can occur intravascular or extravascular. The intravascular mechanisms include direct cellular destruction via lysis, toxins, or trauma; fragmentation and oxidation.
Multiple haemolytic disorders produce substantial intravascular haemolysis. Examples, the corpuscular hemolysis include PNH, extra-corpuscular haemolysis, acquired (autoimmune haemolytic anaemia (AIHA), thrombotic thrombocytopenic purpura (PTT)), as well as other diseases. These disorders are also associated with an increased risk of thrombosis [1-6] (Table 1).
Table 1: Diseases associated with both haemolysis and thrombosis. View Table 1
The clinical presentation of hemolytic anemias is variable and non specific. Haemolysis, which is observed in multiple diseases, can affect all three components of Virchow's triad (hypercoagulability, blood stasis and endothelial injury dysfunction), thus, it is not surprising that there is a link between haemolytic disorders and thrombosis.
Through this work we will try to explain the common pathophysiological mechanisms related to hemolysis, but also the specific mechanisms of each pathology. Of course, the classic risk factors for Thrombotic Diseases are also to take in consideration.
A 28-year-old man was admitted to our institute for evaluation of new tea-colored urine noticed intermittently over the past five days. He also had episodes of headache and dyspnea. On initial evaluation, CBC, urinalysis, and renal ultrasound were normal. A few days later, he developed jaundice with and right calf pain and swelling. He had no history of previous venous thromboembolic disease. Ultrasound revealed occlusive thrombus to right popliteal and right posterior tibial vein.
A Computed Tomography Angiography of the chest was normal. Treatment was started immediately with Lower Molecular Weight and Heparin (LMWH) and compression stockings to which she responded and discharged after 2 weeks with oral anticoagulant. He was followed up for 3 months.
Repeat testing at that time, he was found to have marked anemia Hb 4 g/dL (13-17 g/dL) with high MCV 109 fL (83-101 fL), MCH 34 pg (27-32 pg), and a high reticulocyte percentile of 23% (0.5-2.5%), WBC count 3600/mm3, platelet count 189,000/mm3. Her peripheral smear showed a picture suggestive of hemolysis. However, her renal function was normal. He presented again after 1 year with the same complaints and was found to have marked anemia, with hemolysis again.
The urinalysis showed hemoglobinuria. Direct Coomb test is negative. Flow cytometry of peripheral blood showed absent expression of CD 55 and CD 59 on 80% of red blood cells.
This patient’s clinical presentation is classical for paroxysmal nocturnal hemoglobinuria (PNH). Management approach to our patient includes treatment of anemia and thrombosis. A low transfusional need was maintained. However, due to recurrent episodes of hemolysis and hematuria (LDH progressively increasing to up to 2000 U/L; haptoglobin remained under normal value. A standard-dose eculizumab therapy was started in 2019, consisting of eculizumab 600 mg via 25-45 min IV infusion every week for weeks 1-4, followed by 900 mg IV for the fifth dose 7 days later, then 900 mg IV every 14 days thereafter.
A 43-years-old woman, operated previously for a cholecystectomy and taking combined hormonal contraceptive, were hospitalized for an acute anaemia syndrome with sweelling left inferior extremity. She reported during the last 6 months weekly isolated mild periorbital headaches with blurred vision.
The patient had clinically a focal tenderness with a positive Homans sign without any chest pain or haemoptysis. The neurologic and ophtalmic exams were without abnormality.
The Doppler ultrasound of the inferior extremities showed a deep femoro-popliteal vein thrombosis. She received an intravenous heparin treatment with Vitamin K antagonist therapy.
It was biologically found a mild hyperleukocytosis by a number of white blood cells of 10 600/mm3, a thrombocytopenia of 71,000/mm3, TCA 32/30 seconds, a prothrombin time (TP) of 85%, and a C-reactive protein of 4 mg/L. Renal function, hemoglobin level and the immunological assessment were normal.
The brain scan, the Doppler ultrasound of the supra-aortic trunks, the trans-thoracic echocardiography, the EEG and the pulmonary radiography were all without abnormalities. Antiphospholipid syndrome was initially mentioned but the immunological assessment came back negative.
After one-week, purpuric skin lesion appeared in the ankle, legs and elbows. It was also found a normocytic anemia with a hemoglobin at 6, 6 g/dL and schistocytes were present at the rate of 12%. The platelet count was 13000/mm3; an increased rate of LDHs at 525 UI/L was founded. The haptoglobin was in the opposite decreased to 0.2 g/L and the direct Coombs test was negative. Also a moderate renal impairment was biologically found (urea = 10 mmol/L and serum creatinine = 102 µmol/L).
A thrombotic thrombocytopenic purpura was mentioned and confirmed by the presence of Anti-adamts 13 antibodies against metalloprotease. The diagnosis of a thrombotic thrombocytopenic purpura associated to a deep vein thrombosis was eventually retained.
After 12 plasma exchanges with fresh frozen plasma infusions, the evolution of the patient was conducive. The patient will only present a biological relapse (thrombocytopenia with microangiopathic hemolysis) without clinical symptoms. A rituximab therapy (with a dose of 375 mg/m2 every week through 4 infusions) provided to the patient a two-year prolonged remission.
A 36-year-old man was referred to our department because of a seven-day history of pallor and weakness. The patient's vital signs were stable except for sinus tachycardia of 110 beats/min. His physical examination was remarkable for icterus, hepatosplenomegaly and bilateral lower-extremity. There was no significant peripheral lymphadenopathy.
The complete blood count revealed anemia with a hemoglobin level of 6.5 g/dL, a reticulocyte count of 160000 elmts/mm3 and normal white cell and platelet counts. Hemolysis was confirmed by elevated lactate dehydrogenase (LDH) of 1540 U/L, total bilirubin 3.8 mg/dl, indirect bilirubin 0.9 mg/dl and low haptoglobin of 5.11 mg/dL. Urine examination was negative for hemoglobin and myoglobin. The direct antiglobulin test was strongly positive for IgG autoantibodies. The results of antinuclear antibody and anti-deoxyribonucleic acid were negative. Serum C3 and C4 as well as IgG, IgM, and IgA levels were normal. Serology for cytomegalovirus, Epstein-Barr virus, Mycoplasma pneumonia, and human immunodeficiency virus were negative. Based on the symptoms, the clinical findings and the laboratory tests the warm type of AIHA was established. During the hospital course, the patient was started on intravenous methylprednisolone therapy. When the man was clinically stable, oral prednisolone at a dose of 1 mg/kg/day was then used for 4 weeks followed by a slow taper during the following 5 months, with a good hematological response. One year later, the patient presented with a relapse of the disease (deep anemia at 5 g/dl of hemoglobin) with appearance of a right big leg acute. Her plasma D-dimer level was elevated at 4.20 µg/dL, and doppler ultrasound scanning revealed a thrombosis in her right popliteal vein. Anticoagulant therapy was started. Our patient was placed on corticosteroid and rituximab therapy at a dose of 375 mg/m2 every week. A total of four doses were administered over a period of four weeks. After the first two courses of rituximab therapy, the patient showed a marked clinical improvement.
Many factors can contribute to the risk of venous thrombosis observed in hemolytic diseases. Some mechanisms are related to hemolysis by itself, while others seem more specific to each disease.
The mechanism through which haemolysis leads to thrombosis is multifaceted [7] (Figure 1). Circulating free haemoglobin and haem can have many deleterious effects. In fusion of haem into humans is associated with disturbances in coagulation and thrombophlebitis [8].
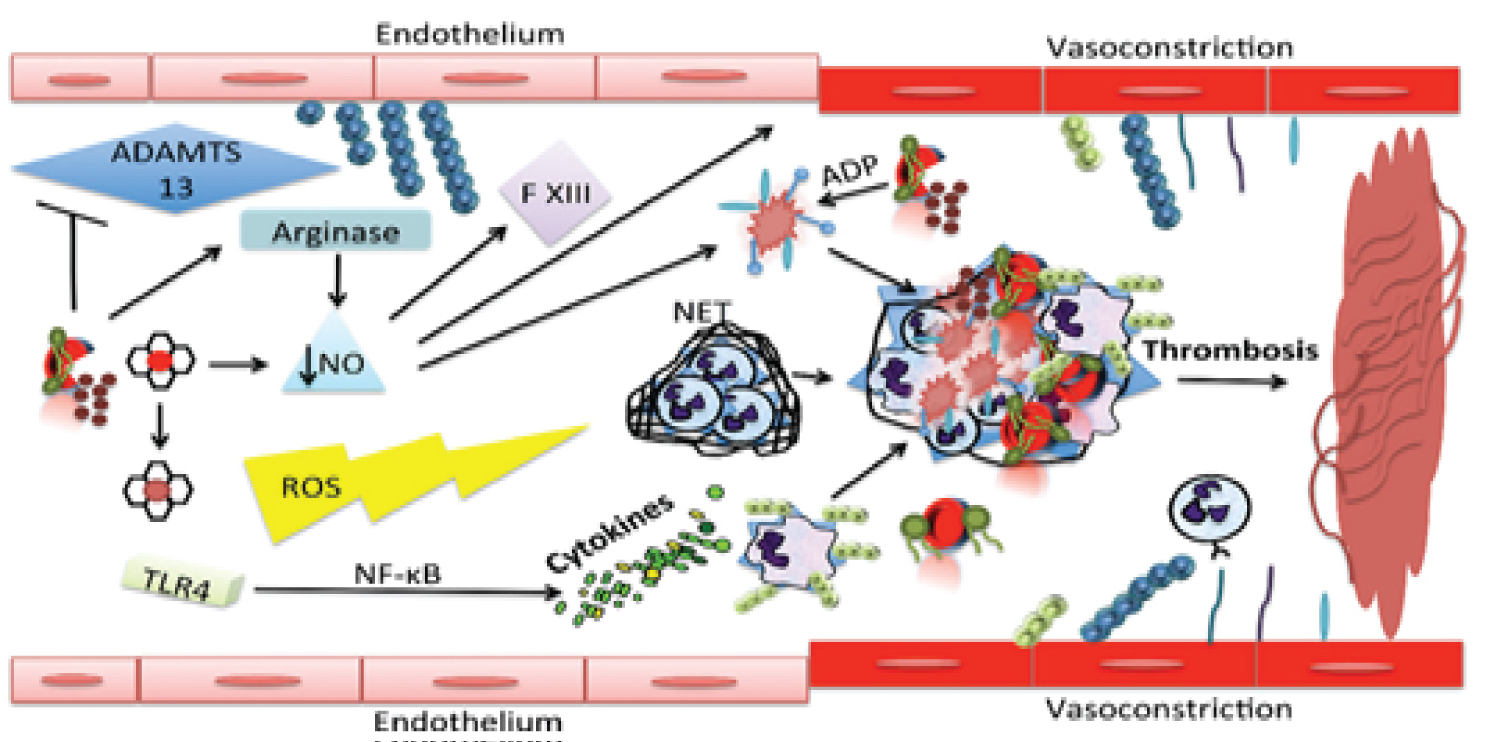 Figure 1: The mechanism through which haemolysis leads to thrombosis is multifaceted.
Figure 1: The mechanism through which haemolysis leads to thrombosis is multifaceted.
RBCs release haemoglobin and arginase into the circulation, which scavenge nitric oxide (NO) and reduce its production, respectively. NO depletion results in vasoconstriction, endothelial cell activation, activation of Factor XIII, and platelet activation and aggregation. Haemolysed RBCs inhibit the function of A Disintegrin and Metalloproteinase with a ThromboSpondin type 1 motif, member 13 (ADAMTS13) and increase circulating microvesicles and PS exposure, stimulating coagulation. ADP is also released, thus activating platelets. Free haem activates neutrophils to form neutrophil extracellular traps (NETs), which recruit RBCs, activate platelets and promote fibrin deposition. Furthermore, the products of haemolysis produce reactive oxygen species (ROS) and induce pro-inflammatory cytokine responses (some mediated by toll-like receptor 4 [TLR4] and nuclear factor-κB [NF-κB]). All of these effects act in concert to increase the risk of thrombosis.
View Figure 1
The main mediator of these adverse effects is thought to be free haem via its effects on NO scavenging, pro-inflammatory cytokine responses, and reactive oxygen species (ROS) generation.
Thus, haemolysis results in NO scavenging, systemic vasoconstriction and increased blood stasis, there by affecting one of the principle components of Virchow’s Triad. Furthermore, Reactive oxygen species (ROS) induction by haem directly, oxidize cell membrane constituents to induce cytotoxicity and promote inflammation and thrombosis.
Furthermore, Immune activation and cytokine secretion depends on the coordinated iron in the porphyrin ring [9], and nuclear factor activation and ROS are essential for the increase in cytokine production induced by haem [10]. Finally, the cytokines produced through these pathways can then affect endothelial activation, leucocyte recruitment, and ultimately thrombotic risk. Platelets may be activated during haemolysis by several different mechanisms involving decreased NO levels, increased pro-inflammatory cytokines and mediators, and ROS. Vascular endothelium has a critical role in maintaining vessel integrity, and as such, has the ability to be both pro- and anti-thrombotic depending on circumstances. Incubating endothelial cells with haem induces tissue factor, which can initiate coagulation [11].
Haem affects neutrophils in multiple ways. DNA component of neutrophil extracellular traps (NETs) also enhances thrombin generation via the intrinsic coagulation pathway [12]. Thus, NETs recruit RBCs, activate platelets and promote fibrin deposition, appear to contribute to the pathogenesis of deep venous thrombosis.
Presenting clinical features of acquired TTP are diverse; some patients have minimal abnormalities, whereas others are critically ill [13]. Weakness, gastrointestinal symptoms, purpura, and transient focal neurologic abnormalities are common. Most patients have normal or only transient, mildly elevated creatinine values. Diagnostic criteria are the presence of microangiopathic hemolytic anemia and thrombocytopenia without another apparent cause. An ADAMTS13 level indicating less than 10% of normal activity supports the clinical diagnosis of acquired TTP. It identifies almost all patients at risk for relapse, but this level is neither sufficiently sensitive to identify all patients with TTP nor sufficiently specific to exclude patients with underlying disorders [14,15].
Treatment for TTP is an emergency, given the frequency of visceral suffering in the acute phase. Treatment is currently based on plasma exchanges which allow the supply of large volumes of plasma (and therefore ADAMTS13). Corticosteroid therapy with methylprednisolone (1 mg/kg/day for 3 weeks with progressive decrease) should be discussed, in the absence of a progressive infection.
Paroxysmal nocturnal hemoglobinuria (PNH) is an acquired disorder characterized by hemolysis, thrombosis, and bone marrow failure caused by mutation in the PIGA (glycosyl phosphatidyl inositol-anchored). PIGA mutations lead to a deficiency of GPI-anchored proteins, such as decay-accelerating factor (CD55) and CD59, both complement inhibitors.
The absence of CD55 and CD59 on blood cells leads to complement-mediated hemolysis and a propensity for thrombosis. Most often diagnosed in early adulthood, PNH affects both sexes equally [16]. One of the signature symptoms of PNH is dark colored urine, but only about 25% of patients present with this symptom. There is an intrinsic relationship between the coagulation cascade and the complement system that is revealed by understanding some of the mechanisms thought to result in thrombosis in PNH. Platelet activation, complement-mediated hemolysis, impaired nitric oxide (NO) bioavailability, impairment of the fibrinolytic system, and inflammatory mediators are all proposed mechanisms and thought to be responsible for the increased thrombotic risk in patients with PNH. The pathogen can also further induce complement activation.
Therefore, thrombosis activates complement, perhaps leading to further thrombosis in PNH and then a vicious circle ensues. Anticoagulation therapy with heparin or low-molecular-mass heparin is still the first action to take in the setting of a new thrombotic event. Complement inhibition therapy with eculizumab should be commenced within 24 hours of any new thrombotic event, wherever possible, to reduce the risks of thrombotic propagation, recurrence and subsequent long-term complications [17].
Autoimmune hemolytic anemias (AIHAs) are rare and heterogeneous disorders characterized by the destruction of red blood cells (RBC). The diagnosis of AIHA is usually simple and based on the presence of hemolytic anemia and serological evidence of anti-RBC autoantibodies detected by the direct antiglobulin test. During AIHA, the thrombotic risk is reported to not correlate with traditional risk factors [18]. Overall, in such a context of AIHA, nor the presence of icterus, the level of bilirubin, the level of LDH, reticulocyte count, nor the level of hemoglobin at AIHA diagnosis or during follow-up appeared as clinically relevant parameters to screen for patients with the higher risk of thrombosis. More recently, it has been shown that haem participates to granulocyte migration and to the release of neutrophil extracellular traps (NET) that could contribute to VTE by recruiting platelets and increasing their activation and thrombin generation [19].
The presence of a lupus anticoagulant identified AIHA patients at higher risk for venous thromboembolism. The favoring role of antiphospholipid antibodies has been reported in several studies [5,20]. Splenectomy has also been reported as an independent risk factor for thrombosis during AIHA [21].
Without criteria to identify the patients with higher risk, we do think that a systematic clinical screening for VTE must be performed at AIHA diagnosis.
The link between hemolysis and venous thrombosis is multifactorial, and involves prothrombotic specific mechanisms related to hemolysis and classic Thrombotic risk factors. Pathological haemolysis overwhelms the physiological defences against the toxicity of iron from the haem group of haemoglobin. The pro‐oxidant, pro‐inflammatory and NO scavenging capacity of free haem affects the three elements of Virchow's triad by causing vasoconstriction, activating coagulation and damaging endothelial cells.
The effects of haemolysis will probably affect the physiopathology of thrombosis in various disorders, including autoimmune haemolytic anaemia, thrombotic thrombocytopenic purpura and Paroxysmal nocturnal hemoglobinuria. A better understanding of the toxic effects of haemolysis will result in better therapies to prevent the side effect of thrombosis.
The authors wish to thank all our participants and resident doctors in the Department of Internal Medicine A, Mohammed V military hospital, Medical school of Rabat, Morocco.
The authors declare no conflict of interest, financial or otherwise.