Aligners are the most important software used in the field of Transcriptomics studies and related fields. In the recent study, almost all the aligners could be configured to give good results, but still, researchers and scientists who use such software face challenges in choosing accurate, sensitive, requiring fewer hardware facilities and ultimately appropriate with their research goals. We try to clarify the various challenges and misunderstandings, below.
Alignment is the first step in most RNA-seq analysis pipelines, and the accuracy of downstream analyses depends heavily on it. Many algorithms have been developed for this alignment step. Due to the increasing growth in the use of aligning and mapping software, this software has become particularly important. This seemingly worthless issue but it is confusing and difficult to compare results from different approaches. We performed a comprehensive benchmarking of 4 popular and common aligners and compared default with optimized parameters. Another thing that should be considered is how robust the results are to different parameters. In the previous studies, almost all the aligners could be configured to give good results, but they differed in the performance of the default options [1], with HISAT2 (hierarchical indexing for spliced alignment of transcripts 2) looking pretty good in those terms [2]. We have to say though, we use HISAT2 a lot just because of how easy it is and how few resources it requires. Therefore, in this research, we have done a statistical analysis using SPSS software on simulated (The simulation engine BEERS [3] was used to generate simulated data. Data were generated for human. Each data set consists of 15 million 100-base paired-end strand-specific reads. The genomes used were Homo sapiens hg19. For human data, 30,000 transcript models were chosen at random) and real data in the GEO (Gene Expression Omnibus) database [4] (Supplementary Table 1 shows the accession number, number of samples and related study title of data used in this study that obtained from different experiments (~116 billion reads)). Some studies have shown related software in comparison with other software in terms of sensitivity, precision, run time and memory usage and shown HISAT2 is more acceptable (Supplementary Table 2) but it's not about the number of mapped reads and the power of other software. In this way, the most important of these software include HISAT2, TopHat2 [5] and STAR [6], and the other hand because HISAT2 uses the Bowtie2 [7] implementation, so we compared these four software in terms of mapping percentage averages (Supplementary Table 3).
Supplementary Table 1: The accession number, number of samples and related study title of data used in this study that obtained from different experiments (~116 billion reads). View Supplementary Table 1
Supplementary Table 2: Comparison of studies. View Supplementary Table 2
Supplementary Table 3: Comparison of four software in terms of mapping percentage averages. View Supplementary Table 3
The simplified results of the comparisons are presented in Table 1. Table 2 shows the percentages of mapping on the human reference genome using Trimmomatic software [8] output files and then using HISAT2, TopHat2, STAR and Bowtie2 aligner (existed aligner in the galaxy server). Reads declared ‘aligned' can be summarized in three main groups: Correctly mapped, correctly multimapped, and incorrectly mapped reads. Hopefully, an effective tool will report the majority of reads aligned correctly, with a few reads aligned ambiguously and very few reads aligned incorrectly (Figure 1).
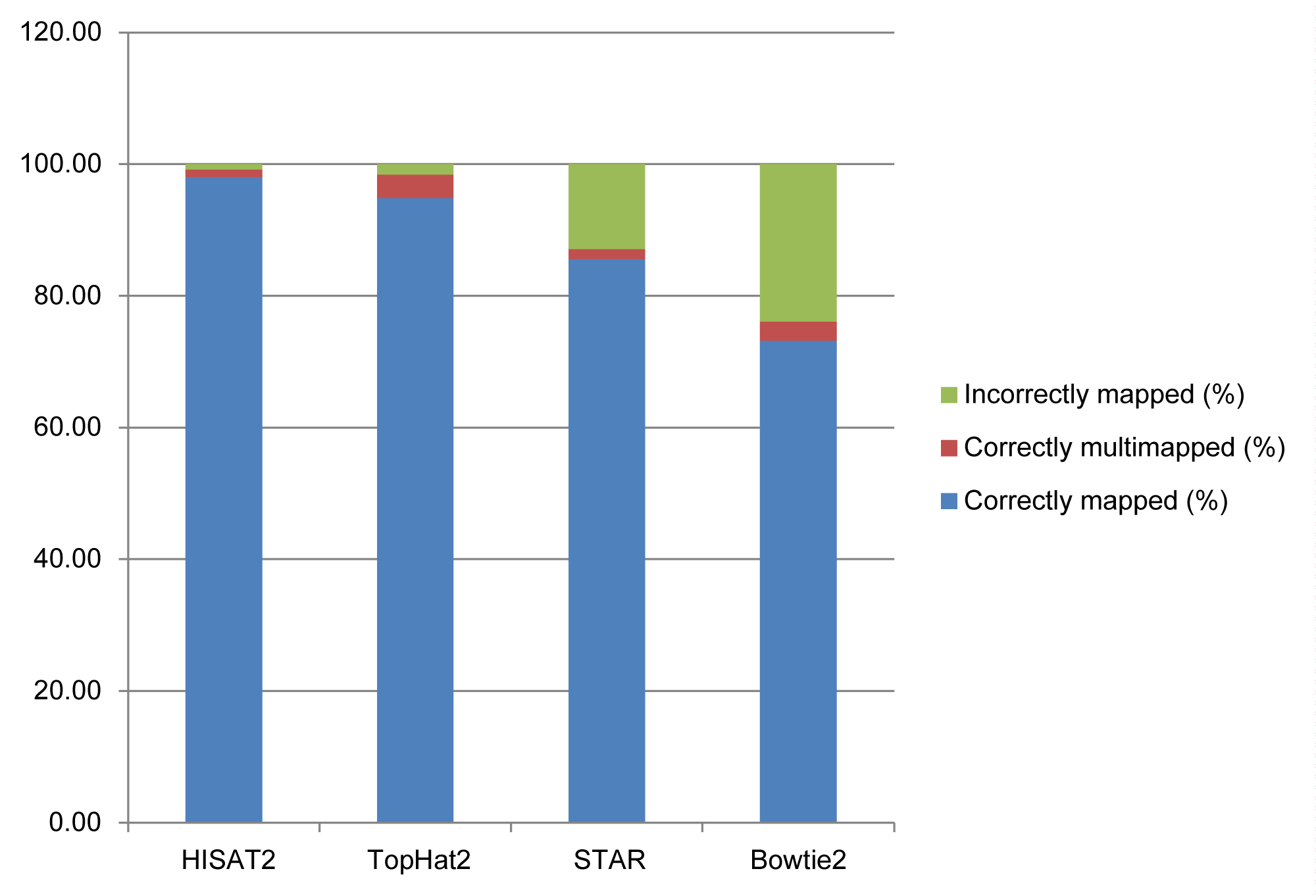 Figure 1: Alignment accuracy of spliced alignment software for 30 million simulated 100-bp reads. Reads are categorized as indicated by the colors. For multimapped reads, an aligner was credited with a correct alignment if it mapped a read to multiple locations. Note that the set of multimapped reads reported by the various aligners may be different, depending on each program's alignment policy and default behavior. The upper numbers are the percentages corresponding to correctly mapped reads. The numbers inside parentheses show percentages for cases correctly mapped and correctly multimapped.
View Figure 1
Figure 1: Alignment accuracy of spliced alignment software for 30 million simulated 100-bp reads. Reads are categorized as indicated by the colors. For multimapped reads, an aligner was credited with a correct alignment if it mapped a read to multiple locations. Note that the set of multimapped reads reported by the various aligners may be different, depending on each program's alignment policy and default behavior. The upper numbers are the percentages corresponding to correctly mapped reads. The numbers inside parentheses show percentages for cases correctly mapped and correctly multimapped.
View Figure 1
Table 1: Sensitivity, precision, run times and memory usage of leading spliced aligners. View Table 1
Table 2: Results of the comparisons of leading spliced aligners. View Table 2
Results of analysis as follows:
• TopHat2 maps a greater percentage of reads on the reference genome. As well as, correctly multimapped percentage is higher than other software; this can be useful in capturing non-coding regions such as miRNAs and other non-coding RNAs (Supplementary Figure1).
• The precision of the HISAT2 is higher when considering the mapped percentage parameter on a particular location.
• On the other hand, TopHat2 has the power to detect introns from exons and map more reads to more than one specific location.
• STAR maps a greater percentage of reads as incorrectly mapped.
• Finally, Bowtie2, which is more specific to DNA-Seq data, is not practical for using in RNA-Seq mapping studies.
For data science, the software must be provided via an easy to use, unified interface, such that they can be easily deployed and sustainably managed. With an understanding of its ability to analyze data set, the researchers will have a better interpretation of their results. Eventually, the results of the statistical analysis of this research can be a good guide for researchers using this software.
The authors have no financial conflicts of interest.
The authors declare that data supporting the findings of this study are available within the article and its Additional files.
We would like to thank Prof. Reza Miraie-Ashtiani and Prof. Mostafa Sadeghi for providing critical feedback and reagents, Ghorban Elyasi Zarringhabaie for technical assistance and the University of Tehran, Department of Animal Science. We also thank the anonymous reviewers for their constructive feedback which significantly improved the quality of our analysis.