Previously, we have shown that StemBell therapy (i.e. mesenchymal stem cell-microbubble complexes subjected to ultrasound) reduced plaque inflammation and plaque destabilization in the aortic root, and thereby inhibited atherosclerosis exacerbation after myocardial infarction (MI). MI also has an effect on heart valves as it increases valve thickness and remodeling. Moreover, hemodynamic disturbances following MI have also been suggested to affect aortic valve (AV) pathology. Therefore, we have analyzed StemBell therapy on AV after MI. Therefore, we have analyzed StemBell therapy on AV after MI.
In atherosclerotic Apolipoprotein E-deficient mice that were fed a high-fat Western diet, MI was induced via permanent left coronary artery ligation. Six days post-MI, the mice received either 5 × 105/100 μL StemBells or vehicle intravenously. The effects of StemBell treatment on the AV were determined 28 days post-MI via (immuno)histochemical analyses.
In all mice, we observed morphological signs of AV sclerosis including thickened valve leaflets, acellularity, glycosaminoglycan (GAG) transformation and fibrosis, but without evidence of mineralization, suggestive for more early-stage AV disease in our mouse model. StemBell therapy resulted in a significantly decreased infiltration of Mac3+ macrophages in the AV, without significantly affecting CD163+ macrophages, CD45+ leukocytes and monocytic chemoattractants. The advanced glycation end products (AGEs) Nε-(carboxymethyl) lysine (CML) and methylglyoxal (MGO) were decreased in the AV of StemBell-treated mice, albeit not significantly. No significant effects were observed in AV thickness, fibrosis, GAGs, lipid- and calcium-deposits. StemBell therapy did however increase AV iron deposits, although not significant.
StemBell therapy reduced macrophage infiltration of AV after MI without changing valve morphology.
Aortic valve stenosis, Atherosclerosis, Myocardial infarction, Stem cells, StemBell therapy
AV: Aortic Valve; AVR: Aortic Valve Replacement; MI: Myocardial Infarction; MV: Mitral Valve; LV: Left Ventricle; MSC: Mesenchymal Stem Cell; ASCs: Adipose tissue-derived Mesencymal Stem Cells; ICAM-1: Intracellular Adhesion Molecule-1; ILH: Intra-Leaflet Hemorrhage; EvG: Elastica van Gieson; HRP: Horseradish Peroxidase; GAG: Glycosaminoglycan; AGE: Advanced Glycation End; CML: Nε-(carboxymethyl) lysine; MGO: Methylglyoxal; VICs: Valve Interstitial Cells; RAGE: Receptor for Advanced Glycation End; ECM: Extracellular Matrix
Aortic Valve (AV) stenosis is the most common type of valvular disease in the Western world [1]. AV stenosis occurs mainly in the elderly population with an increasing prevalence of critical aortic stenosis from 1-2% in individuals aged 75/76 years to 6% in those aged 85/86 years [2]. The spectrum of AV stenosis varies from early lesions with mild leaflet thickening and calcification without obstruction, also called AV sclerosis, to severe calcified AV stenosis with obstruction [3]. Currently, the only effective treatment available is AV replacement (AVR) via either a surgical or percutaneous approach, since no other medication-based therapies have been shown to alter the poor prognosis of severe AV stenosis [2]. However, not all patients are eligible for AVR due to the risk of complications, pointing to the importance of developing new effective alternative therapies for AV stenosis [4].
The underlying pathophysiological mechanisms of AV sclerosis and eventual stenosis are still poorly understood, but recent studies indicate that sclerotic AV leaflets have many features similar to atherosclerotic plaques of arteries, e.g. lipid accumulation and calcification [1]. Furthermore, both sclerotic AVs and atherosclerotic plaques of arteries are characterized by inflammation [5], which involves infiltration of CD45+ leukocytes, CD68+ macrophages and CD3+ T-cells, fibrosis, increased vascularization and increased valve weight [6]. Moreover, a higher transaortic peak gradient was observed in patients with inflammatory infiltrates in their AV compared to patients without intra-valvular inflammatory infiltrates [6]. Taken together, these findings suggest that infiltration of inflammatory cells in the heart valve is an important contributor in the pathobiology of AV stenosis. Therefore, inhibiting infiltration of inflammatory cells might be a promising therapeutic approach.
Previously, it was observed that myocardial infarction (MI) in humans was associated with increased atherosclerotic inflammation of the aorta [7]. Moreover, MI induced by coronary artery ligation in ApoE-/- mice caused a systemic inflammatory response accompanied by macrophage mobilization, increased atherosclerotic inflammation and accelerated plaque progression of the aortic root [8]. These findings implicate that MI itself exacerbates systemic atherosclerotic inflammation triggering plaque destabilization. Interestingly, MI has been shown to increase mitral valve (MV) thickness and remodeling [9]. Moreover, it is known that reduced left ventricle (LV) contractility - which often follows upon MI due to loss of contractile tissue within the myocardium [10] - leads to slower opening of the AV and a smaller orifice area [11]. Subsequently, this causes a disturbance in the pressure gradient across the AV and the transvalvular flow [12], which all have been suggested to affect AV pathology [13,14]. These results thus point to a jeopardizing role for MI in AV disease.
Previously, it was shown that mesenchymal stem cell (MSC) therapy is effective in tissue regeneration in cardiovascular disease [15,16], especially related to the immunomodulatory and anti-inflammatory effects, as they induced macrophage polarization and enhanced anti-inflammatory cytokines secretion [17,18]. Recently, we developed a new method to enhance the efficacy of MSC therapy, the so called 'StemBell technology' [19]. StemBells are adipose tissue-derived MSCs (ASCs) coupled to ultrasound contrast microbubbles that are covered with antibodies against the ASC marker CD90 and against the target molecule intracellular adhesion molecule-1 (ICAM-1), which is expressed on activated endothelial cells in atherosclerotic plaques [20-22]. These StemBells are susceptible to the acoustic radiation force of ultrasound and therefore can be guided in vivo to the desired location by applying local ultrasound [19,23]. We have shown that this technology targeted ACS to the infarction area in rats after MI and induced a long term improvement of the cardiac function [24,19]. In addition, we have shown recently that StemBell therapy increased cap thickness and reduced intra-plaque inflammation in atherosclerotic plaques of the aortic root after MI in atherosclerotic ApoE-/- mice [23].
Since sclerotic AVs share many features with atherosclerotic plaques, including enhanced ICAM-1 expression, we wondered whether StemBell therapy would also have a protective effect on the AV after MI by analyzing the AV obtained from the previously performed mouse experiment [23].
We analyzed aortic valves (AVs) isolated from a previously performed animal experiment [23]. Female, 6-week-old ApoE-/- on a C57BL/6 genetic background (n = 26; Taconic, Denmark) were fed a high-fat diet (0.15% cholesterol; 4022.83, AB Diets, Woerden, The Netherlands) for 10 weeks to induce atherosclerosis. Blood samples were collected via the tail vein upon a 4 hour fasting period after 7 weeks of diet. Upon 10 weeks of diet, MI was induced via permanent left coronary artery ligation as described previously [23]. Subsequently, the animals were randomly assigned to the vehicle (n = 13) or the StemBell (n = 13) group.
StemBells were assembled by incubating the ASCs that were obtained from the adipose tissue of the abdominal subcutis and inguinal fat pads of 12-week-old female wild type C57Bl6 mice (n = 13; Charles River, The Netherlands), with the dual-targeted microbubbles in a 1:100 cell:microbubble ratio at a concentration of 5 × 105 StemBells per 100 µl in serum-free MesenCult medium under continuous rotation for 30 minutes at room temperature.
Six days after MI, either 100 µl Complete MesenCult Medium (vehicle) or freshly prepared 5 × 105 StemBells (in 100 µl Complete MesenCult Medium) were injected in the tail vein. Thereafter, 1 MHz transthoracal ultrasound was applied with an acoustic pressure of 100 kPa and a 1 kHz pulse repetition frequency to all animals by positioning the transducer (V303-SU, Panametrics Inc, Waltham, MA, USA) parasternal directed at the anterior wall of the heart for 60 seconds. The transducer was coupled to a waveform generator (33220A, Agilent, Palto Alto, CA, USA) and a linear 60-dB power amplifier (150A100B, Amplifier Research, Bothell, WA, USA). Twenty-eight days after MI induction, again blood samples were collected via the tail vein after a 4 hour fasting period. Then, mice were sedated under 5% isoflurane anesthesia, followed by collecting blood via cardiac puncture using ethylenediaminetetraacetic acid (EDTA) solution (Ambion AM9260G, 7.7 × 10-2 molar) as an anti-coagulant. Immediately after the blood collection the mice were sacrificed. For this study, dissected heart segments containing the aortic root together with the AV leaflets, were used for (immuno)histochemical analysis.
This study was approved by the animal ethics committee of the Amsterdam UMC – location VUmc (former VU University Medical Center).
The aortic root was cross-sectioned in its entirety (4 μm). After every 200 μm a cross-section was stained with Toluidine Blue 1% reagent (Merck) to determine which cross-sections contained the AVs. The cross-sections were divided over 14 slides. The first cross-section that contained AV tissue was mounted on the first slide. The consecutive cross-section on the second slide, etcetera until slide 14. Cross-section 15 was mounted on slide 1 again and so forth until each of the 14 slides contained 8 cross-sections. This ensured that the complete AV was analyzed and that each staining was performed on multiple cross-sections throughout the AVs.
Sections were stained with Elastica von Gieson (EvG) for detection of fibrosis. Glycosaminoglycans (GAGs) were visualized by Alcian Blue staining. Calcium deposits were visualized by von Kossa staining. Iron deposits, indicative for intra-leaflet hemorrhage (ILH), were identified by the use of Perl's iron staining. Only cross sections with intact AV leaflets or those with the highest amount of tissue for each AV leaflet were elected for analysis.
The average AV thickness was measured on scanned Elastica van Gieson (EvG) stained slides using Panoramic Viewer 1.15.2 software (3DHistech, Budapest, Hungary). The surface area of all three AV leaflets (μm2) was measured and summed. This number was divided by the combined length of the AV (μm) to obtain the average AV thickness (μm). The maximal leaflet thickness of the AV was determined at the nodule of Arantius in the middle of each leaflet (μm). The percentage of the total AV surface area positive for fibrosis and GAGs was determined on scanned EvG and Alcian blue stained slides respectively by use of Image J software via color-gating. All AVs were evaluated for von Kossa and Perl's iron positivity. The percentage of mice with Perl's iron-positive AVs per treatment group (vehicle versus Stembell) was determined. The area positive for lipids was calculated by subtracting the area positive for fibrosis and GAGs from the total AV area.
For immunohistochemical analysis, sections were deparaffinized, rehydrated and blocked for endogenous peroxidases via incubation in 0.3% H2O2 diluted in methanol for 30 minutes. Antigen retrieval via heat inactivation was then performed by boiling the sections in 10 mM citrate buffer (pH 6.0) for 10 minutes (CD45, Mac3, CD163, MGO) or pepsin was used for 30 minutes at 37 ℃ (CML) or boiling the sections in Tris/EDTA buffer (pH 9.0) for 10 minutes (CX3CL1- and MCP-1). Subsequently, the sections for anti-CD45, anti-Mac3, anti-CML, and anti-MGO-derived AGEs staining were pre-incubated with normal rabbit serum (1:50 DAKO X0902) for 10 minutes. For anti-CD163 staining, sections were pre-incubated with normal swine serum (1:20, Jackson, 014000121). Then, sections for CD45, Mac3, CD163 staining were incubated with monoclonal rat anti-mouse CD45 (1:50, BD, Bioscience, 550539), or monoclonal rat anti-mouse Mac3 (1:30, DB, Pharmingen, 55332), or polyclonal rabbit anti-mouse CD163 (1:100, Bioconnect, bs-2527R), or rat-anti-mouse MCP-1 (1:10, Acris, AM26342PU-N) overnight. Sections for CX3CL-1, CML and MGO staining were incubated with rabbit-anti-mouse CX3CL-1 (1:10, Cloud cone crop, RPA040Mu01), or anti-CML26 biotinylated antibody (1:200, homemade [25]), or anti-MGO-derived AGEs (1:25, homemade [26]) for 60 minutes respectively. Thereafter, the sections were incubated for 30 minutes with the secondary antibodies rabbit anti-rat-HRP (1:50, Dako, P050) for CD45 and Mac3 staining, swine anti-rabbit horseradish peroxidase (HRP) (1:200, Dako, P0217) for CD163 macrophage and CX3CL-1 staining, rabbit-anti-rat HRP (1:200, Abcam, 6734) for MCP-1 staining, and rabbit-anti-mouse biotinylated antibody (1:500, Dako, E0413) for CML and MGO staining. Sections for CML and MGO were incubated with the third antibody streptavidine-HRP (1:200, Dako, P0397) for 60 minutes. All stainings were visualized via incubation in diaminobenzidine (Dako, K3468) for 10 minutes. The sections were then counterstained with haematoxylin, dehydrated and covered.
The number of leukocytes, macrophages, M2 macrophages per mm2 was determined by dividing the number of intra-AV CD45+, MAC3+, CD163+ (counted using a light microscope; Zeiss; Germany) by the surface area of the corresponding AV. The percentage of the total AV area positive for CXC3L-1 and MCP-1 was measured on scanned CX3CL-1 and MCP-1 stained slide using Panoramic Viewer 1.15.2 software (3DHistech, Budapest, Hungary), while the percentage AV area positive for CML and MGO was determined on scanned CML and MGO stained slides using Image J software via color-gating. The ratio of positive staining area to AV area was calculated (%).
Statistical analyses were performed using Graphpad Prism software, version 7.0 (La Jolla, CA, USA). To evaluate for differences in average AV thickness, maximal leaflet thickness, fibrosis, GAGs, CML, MGO, inflammatory cells and chemoattractants an independent sample t-test was used when data were normally distributed, or a Mann-Whitney U test if not. Results were considered statistically significant if the two-sided p-value was < 0.05. To test for difference in the presence of iron deposits between the vehicle and StemBell therapy group, a Fisher exact test was performed. Numeral data in the text and figures are represented as mean ± SD.
In all mice, we observed morphological signs of AV sclerosis including thickened valve leaflets, acellularity, glycosaminoglycan (GAG) transformation and fibrosis in line with previous observations in atherosclerotic mouse models [27,28]. However, we were not able to detect evidence of mineralization by von Kossa staining in AV leaflets of all the mice, implicating that end-stage calcification is limited in our mouse model and instead corresponds more to early-stage AV disease.
The number of valvular Mac3+ macrophages was markedly and significantly decreased in StemBell-treated mice (9.7 ± 17.8 cells/mm2) compared to vehicle-treated mice (60.9 ± 73.0 cells/mm2; p = 0.016; Figure 1A). This corresponded with a non-significant increase in the number of anti-inflammatory CD163+ macrophages in the StemBell-treated group (113.1 ± 180.6 cells/mm2) compared to the vehicle group (35.1 ± 66.6 cells/mm2; Figure 1B). In addition, a non-significant increase in the number of CD45+ leukocytes was noted in the StemBell group (81.2 ± 76.0 cells/mm2) compared to the vehicle group (56.1 ± 4.8 cells/mm2; Figure 1C).
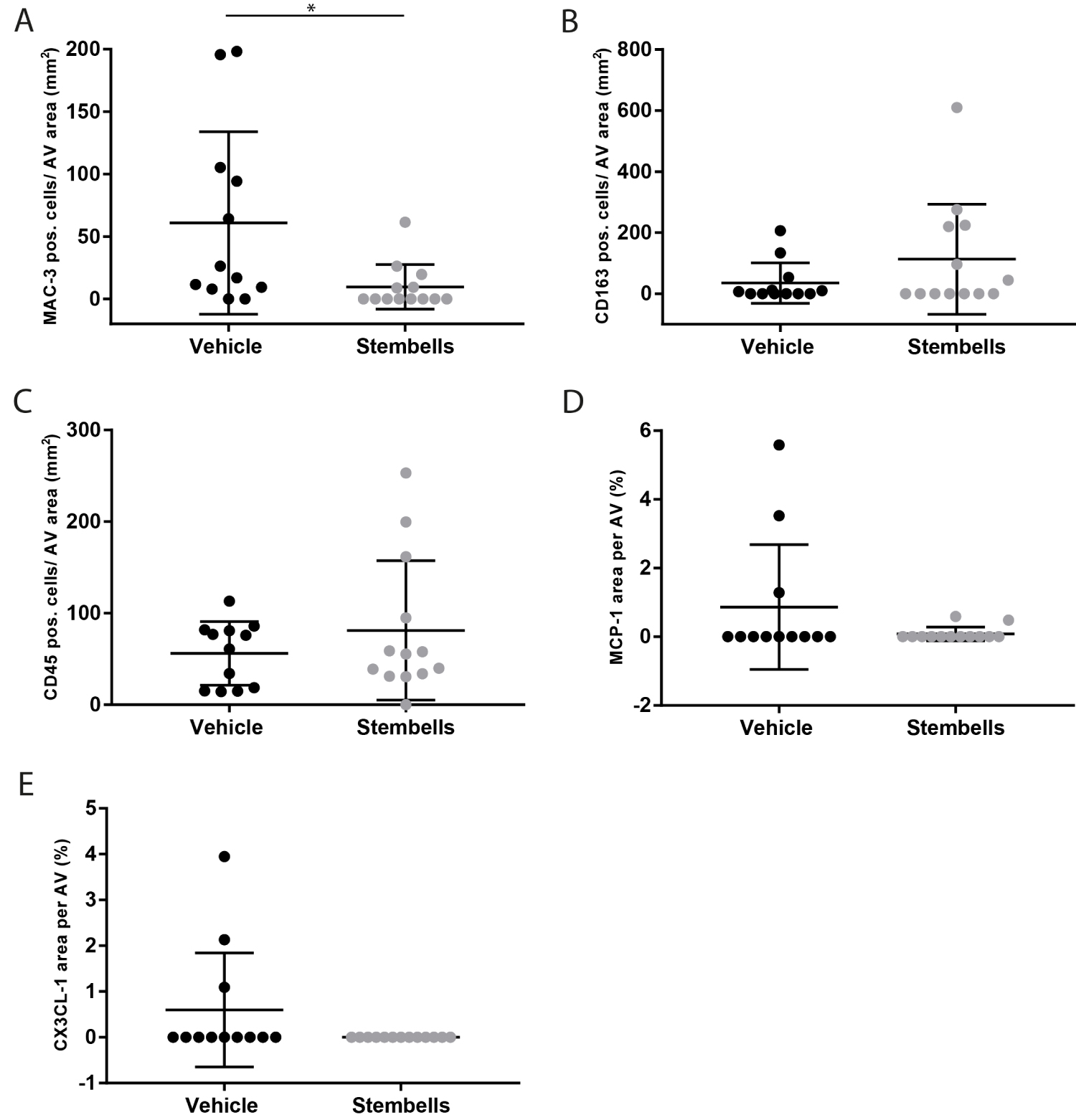 Figure 1: Effect of StemBell therapy on inflammation of the AV: Atherosclerotic ApoE-/- mice underwent myocardial infarction and received either 5 × 105 StemBells or vehicle intravenously; (A) MAC-3 positive macrophages in the AV divided by total surface area of the AV (cells/mm2); (B) CD163 positive macrophages (anti-inflammatory type) in the AV divided by total surface area of the AV (cells/mm2); (C) CD45 positive leukocytes in the AV divided by total surface area of the AV (cells/mm2); (D) Percentage of the MCP-1 positive area in the AV; (E) Percentage of the CX3CL-1 positive area in the AV. Values are mean ± SD, *p < 0.05.
View Figure 1
Figure 1: Effect of StemBell therapy on inflammation of the AV: Atherosclerotic ApoE-/- mice underwent myocardial infarction and received either 5 × 105 StemBells or vehicle intravenously; (A) MAC-3 positive macrophages in the AV divided by total surface area of the AV (cells/mm2); (B) CD163 positive macrophages (anti-inflammatory type) in the AV divided by total surface area of the AV (cells/mm2); (C) CD45 positive leukocytes in the AV divided by total surface area of the AV (cells/mm2); (D) Percentage of the MCP-1 positive area in the AV; (E) Percentage of the CX3CL-1 positive area in the AV. Values are mean ± SD, *p < 0.05.
View Figure 1
Previously, we observed that the StemBell therapy-induced decrease in macrophages was accompanied by a decrease of MCP-1 (a chemoattractant for pro-inflammatory classical monocytes) and an increase of CX3CL-1 (a chemoattractant for inflammation resolving non-classical monocytes) in atherosclerotic plaques of the aorta [23]. Although the average levels of both chemoattractants appeared lower in the AV of the StemBell group, no significant differences were observed between StemBell-treated mice and vehicle-treated mice respectively in the levels of MCP-1 (0.08 ± 0.2% versus 0.87 ± 1.8%; Figure 1D) and CX3CL-1 (0 ± 0% versus 0.60 ± 1.2%; Figure 1C).
Thus, overall StemBell therapy led to significantly decreased numbers of macrophages in the aortic valves without significantly affecting leukocytes and anti-inflammatory macrophages.
In the past we have shown increased presence of the advanced glycation end product (AGE) Nε-(carboxymethyl) lysine (CML) in human diseased aortic valves that correlated with inflammation [25]. For this we have analyzed whether StemBell therapy affected CML and the precursor of AGEs methylglyoxal (MGO) in the sclerotic AVs.
Both CML and MGO were diffusely present within AV leaflets. The percentages of CML and MGO positive area were somewhat decreased in the AV upon StemBell therapy (54.3 ± 18.1% and 22.7 ± 15.3% respectively) when compared to vehicle-treated mice (65.5 ± 17.2% and 30.9 ± 22.4% respectively), however not significantly (Figure 2A and Figure 2B).
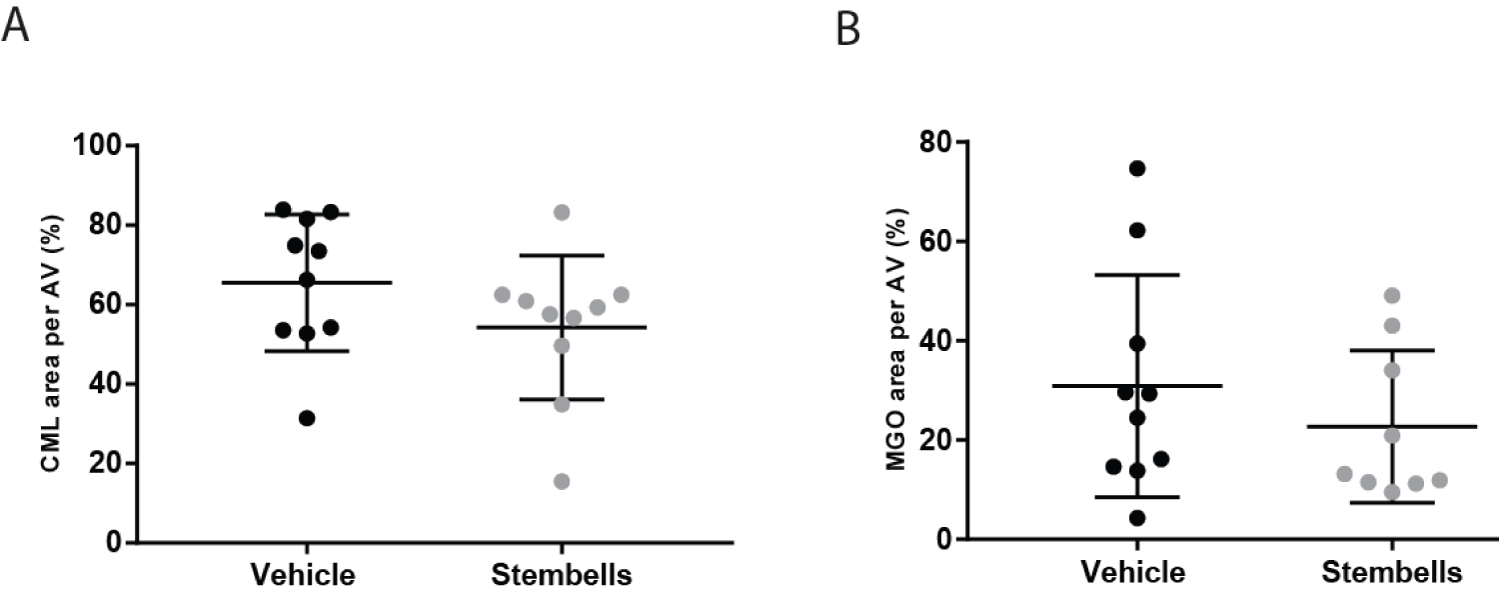 Figure 2: Effect of StemBell therapy on Advanced Glycation Endproducts in the AV; (A) The percentage of CML positivity of the AV is displayed per group; (B) The percentage of MGO positivity in the AV is displayed per group. Arrows indicate positive staining for CML (above) and MGO (below) in an ApoE-/- mouse. Values are mean ± SD.
View Figure 2
Figure 2: Effect of StemBell therapy on Advanced Glycation Endproducts in the AV; (A) The percentage of CML positivity of the AV is displayed per group; (B) The percentage of MGO positivity in the AV is displayed per group. Arrows indicate positive staining for CML (above) and MGO (below) in an ApoE-/- mouse. Values are mean ± SD.
View Figure 2
Thickening of AV, fibrosis and increased depositions of glycosaminoglycans (GAGs), lipids and calcium are all features of AV remodeling [29-33].
The average AV thickness between StemBell (155.9 ± 54.1 μm) and vehicle (158.4 ± 51.8 μm) treated mice however were similar (Figure 3A). Moreover, no effects on maximal leaflet thickness were observed between vehicle- (362.3 μm ± 106.1) and StemBell-treated mice (354.9 ± 105.7 μm; Figure 3B). Figure 3C depicts representative EvG cross-sections of the AV from both the vehicle- and StemBell-treated group.
 Figure 3: Stembell therapy does not affect AV morphology; (A) Average AV thickness (μm) and; (B) Maximal leaflet thickness (μm) displayed per group; (C) Representative images of EvG stained AV from both a vehicle- and StemBell-treated mouse; (D) The percentage of fibrosis and; (E) Glycosaminoglycan (GAG) depositions displayed per group. Values are mean ± SD.
View Figure 3
Figure 3: Stembell therapy does not affect AV morphology; (A) Average AV thickness (μm) and; (B) Maximal leaflet thickness (μm) displayed per group; (C) Representative images of EvG stained AV from both a vehicle- and StemBell-treated mouse; (D) The percentage of fibrosis and; (E) Glycosaminoglycan (GAG) depositions displayed per group. Values are mean ± SD.
View Figure 3
Neither were significant differences observed in the fibrotic area (detected via EvG staining) between vehicle- (69.2 ± 8.9%) and StemBell-treated mice (67.3 ± 8.7%; Figure 3D). No significant effects on GAG-positive areas, determined via Alcian Blue staining, were observed between StemBell (69.2 ± 12.4%) and vehicle (72.1 ± 10.1%) treated mice (Figure 3E). No lipids or calcium deposits (determined using a von Kossa staining), were observed in both the vehicle- and Stembell-treated mice (data not shown).
Lastly, a putative effect of StemBell therapy on intra-leaflet hemorrhage (ILH), indicative for a leaky neovascularity [34], was analyzed using a Perl's iron staining. The presence of iron deposits in the AV was higher in the StemBell-treated mice (46.2%) compared to the vehicle-treated mice (33.3%), however this was not significant. Figure 4 depicts a representative Perl's iron staining in an AV leaflet of vehicle- and StemBell-treated mouse, indicative for ILH.
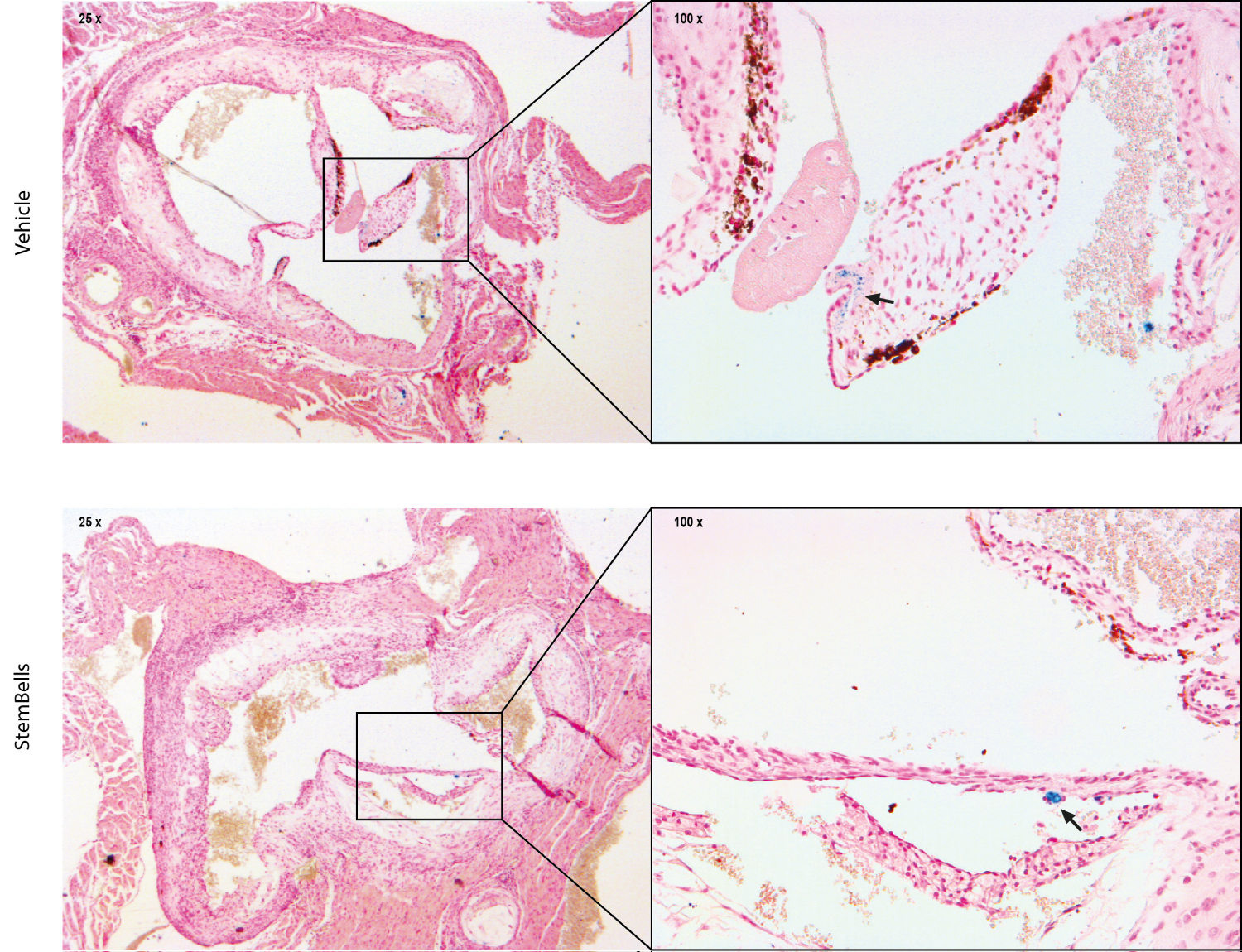 Figure 4: Intraleaflet hemorrhage: Representative images of an AV leaflet positive for Perl's iron staining in a vehicle- and StemBell-treated mouse. Iron deposits are indicated by blue staining (arrow).
View Figure 4
Figure 4: Intraleaflet hemorrhage: Representative images of an AV leaflet positive for Perl's iron staining in a vehicle- and StemBell-treated mouse. Iron deposits are indicated by blue staining (arrow).
View Figure 4
In conclusion, StemBell therapy did not significantly affect AV morphology.
Recently, we showed that systemic StemBell therapy reduced atherosclerotic plaque inflammation and plaque destabilization of the aortic root after MI, which was predominantly associated with local and systemic effects on the number of macrophages/monocytes [23]. We now show that StemBell therapy in atherosclerotic mice with MI also reduced the number of valvular macrophages without affecting other inflammatory cells or valve morphology.
To the best of our knowledge, we are the first to study the use of a stem cell-based therapy as a therapeutic option to prevent or slow down progression of AV sclerosis. In humans and atherosclerosis-prone mouse models, macrophage infiltration was identified as an early event in the development of AV stenosis [35,36]. Moreover, recently it was found that the number of infiltrating CD68+ macrophages was significantly larger in human calcified AVs compared to non-calcified AVs [37]. In addition, it was demonstrated that medium from unstimulated macrophages promoted osteogenic differentiation of valve interstitial cells (VICs) in vitro, suggesting that infiltration of macrophages is in the progression of AV stenosis [37]. Therefore, our observation that StemBell therapy reduced the number intra-valvular macrophages, suggests that StemBell therapy might be able to inhibit AV disease progression by reducing the infiltration of macrophages.
Previous studies have shown that MI induces monocytopoiesis in the spleen, which in turn causes an increase in Ly-6Chigh classical monocyte levels both in the circulation and in atherosclerotic plaques [8]. Previously, we observed that StemBell therapy caused an increase in the percentage of anti-inflammatory CD11b+/Ly-6Clow non-classical monocytes in the blood and a decrease in intra-plaque macrophage content in the atherosclerotic changed aorta 28 days post-MI [23]. This coincided with an increase in the percentage of intra-plaque anti-inflammatory CD163+ macrophages, a decrease in intra-plaque MCP1 levels and an increase in intra-plaque CX3CL-1 levels, chemoattractants for respectively classical and non-classical monocytes [23]. These data suggest that StemBell therapy had an inflammation-resolving effect both locally in the aortic atherosclerotic plaque as well as systemically in the circulation. In the AV next to a significant decrease in intra-valvular Mac3+ macrophages after StemBell therapy we did observe an increased trend in the CD163+ macrophages similar as to our finding in the atherosclerotic plaque, although without reaching statistical significance. Furthermore, the levels of MCP-1 and CX3CL-1 were not significantly affected in the AV. We cannot fully explain these differences, but it is striking that the levels of both MCP-1 and CX3CL-1 in the AV are much lower compared to these levels observed in atherosclerotic plaque of the aortic root [23]. In fact, MCP-1 and CX3CL-1 were absent in the AV of the majority of mice. Given that intra-valvular macrophages were present in most mice, this might implicate that the role of chemoattractants in AV tissue is different and less profound than that observed in atherosclerotic plaque tissue of the aortic root. In addition, we did not find a significant effect of StemBell therapy on the number of intra-valvular CD45+ leukocytes that was even somewhat increased upon StemBell therapy, though not significantly. In line with this, the number of intra-plaque CD45+ leukocytes in the aorta was also not affected upon StemBell therapy [23].
Previously, we have shown that the pro-inflammatory component CML deposition was increased in human degenerated AVs [25]. We now found that StemBell therapy reduced the valvular percentage of two AGEs, namely CML and MGO, although not significantly. Interestingly, it was described that deficiency of the receptor for AGEs (RAGE) in ApoE-/- mice attenuated AV calcification and suppressed macrophage infiltration, suggesting a causative role for AGEs and its receptor RAGE in AV inflammation [38]. Therefore, it is tempting to speculate that the decrease of valvular CML and MGO levels after StemBell therapy might play a role in the decrease of macrophages.
Other important features of AV sclerosis include valve leaflet thickening, extracellular matrix (ECM) remodeling, accumulation of lipids and calcium deposits [29,39]. We however did not find an effect of StemBell therapy on AV thickness, on average AV thickness or on maximal leaflet thickness. We also did not observe an effect of StemBell therapy on fibrosis or GAGs depositions in the AV, both important in ECM remodeling [40]. In the atherosclerotic plaque of the aorta, though, we previously did observe an increase of the fibrous cap in StemBell-treated mice [23]. However, previous studies emphasized that the characteristic fibrous cap on atherosclerotic plaques of the aorta is absent in stenotic AV leaflets. Likewise, we also did not observe a fibrous cap but diffuse fibrosis in the AV leaflets [41]. Furthermore, no significant changes in calcium- and lipid-deposits in the AV were noted between StemBell- and vehicle-treated mice. StemBell therapy did also not affect serum cholesterol- and triglyceride-levels [23].
In the current study, we found limited iron deposits in the AV of both vehicle and StemBell-treated mice. Intra-valvular iron was suggested to originate from intra-leaflet hemorrhages and haem metabolism by macrophages [42,43]. Remarkably, in the current study iron deposits were more often present in StemBell-treated mice than in vehicle-treated mice, albeit not significantly. Since, we also observed significantly less intra-valvular macrophages in the StemBell-treated mice, we cannot adequately explain this increase in intra-valvular iron.
Finally, one could argue that the reduced infiltration of macrophages in the AV might also be explained by reduced hemodynamic changes resulting from StemBell therapy-induced effects on the infarcted heart. Previous studies namely indicated that abnormal hemodynamics - often following MI - experienced by valve leaflets can cause tissue inflammation [10-14]. In the previous study however, we did not observe significant differences in infarct size and cardiac function after StemBell therapy [23] making it less likely for StemBell therapy to influence hemodynamic function in our mouse model.
In summary, we have shown that StemBell therapy reduced macrophages in the AV upon MI without affecting valve morphology. Additional studies are required to fully understand all the observed effects of StemBell therapy for the purpose of adapting their use to treat progressive AV sclerosis in humans.
We acknowledge the kind gift of the biotinylated microbubbles by Prof. Dr. N. de Jong (Department of Biomedical Engineering, Erasmus MC, Rotterdam). This study was funded by ZonNW (Translational Adult Stem Cell Research program, grant number 116005003), by a grant from the Amsterdam Cardiovascular Sciences, The Netherlands, and by Edwards Life Sciences.
All authors declare that they have no conflict of interest.
All authors declare they contributed equally to this manuscript.