Vulvar lymphangitic carcinomatosis (VLC) secondary to gastric carcinoma is a very rare entity. In this paper, we describe the clinical and pathological characteristics, evolution, of a patient with VLC of signet ring cell gastric adenocarcinoma. To our knowledge this is among the very rare reported cases in the literature.
Vulva is an exceptional site for metastasis [1]. Common gynecological or non-gynecological malignancies can be the source of metastasis in vulva [2]. Presence of vulvar metastasis may initially be mistaken as a benign or a primary vulvar malignancy [3]. Differentiating primary and secondary vulvar malignancy is primordial because vulvar metastasis is often a pre-terminal phenomenon [4].
A 32-year-old woman was admitted to the gynecology department to manage a vulvar mass that appeared 3 months ago. The clinical examination found nothing special, the gynecological examination showed a vulvar mass of about 5 cm. A presumptive diagnosis of a benign lesion was made and a surgical decision to remove the mass was made.
Gross examination of the surgical specimen showed a firm gray mass at palpation measuring 5.8 × 3.2 cm (Figure 1). Histologically, a squamous epithelium surmounting a vessel-rich chorion was observed. These vessels contained in their lumen a carcinomatous proliferation made of abundant cytoplasmic cohesive cells, pushing the nucleus in the periphery and producing the appearance of signet ring cells (Figure 2 and Figure 3). The immunoprofile of intravascular tumor cells showed a strong positivity for AE1/AE3 and a moderate positivity for CK7. CD 34 was positive in vessels (Figure 4, Figure 5 and Figure 6).
 Figure 1: Gross examination showing a gray firm mass.
View Figure 1
Figure 1: Gross examination showing a gray firm mass.
View Figure 1
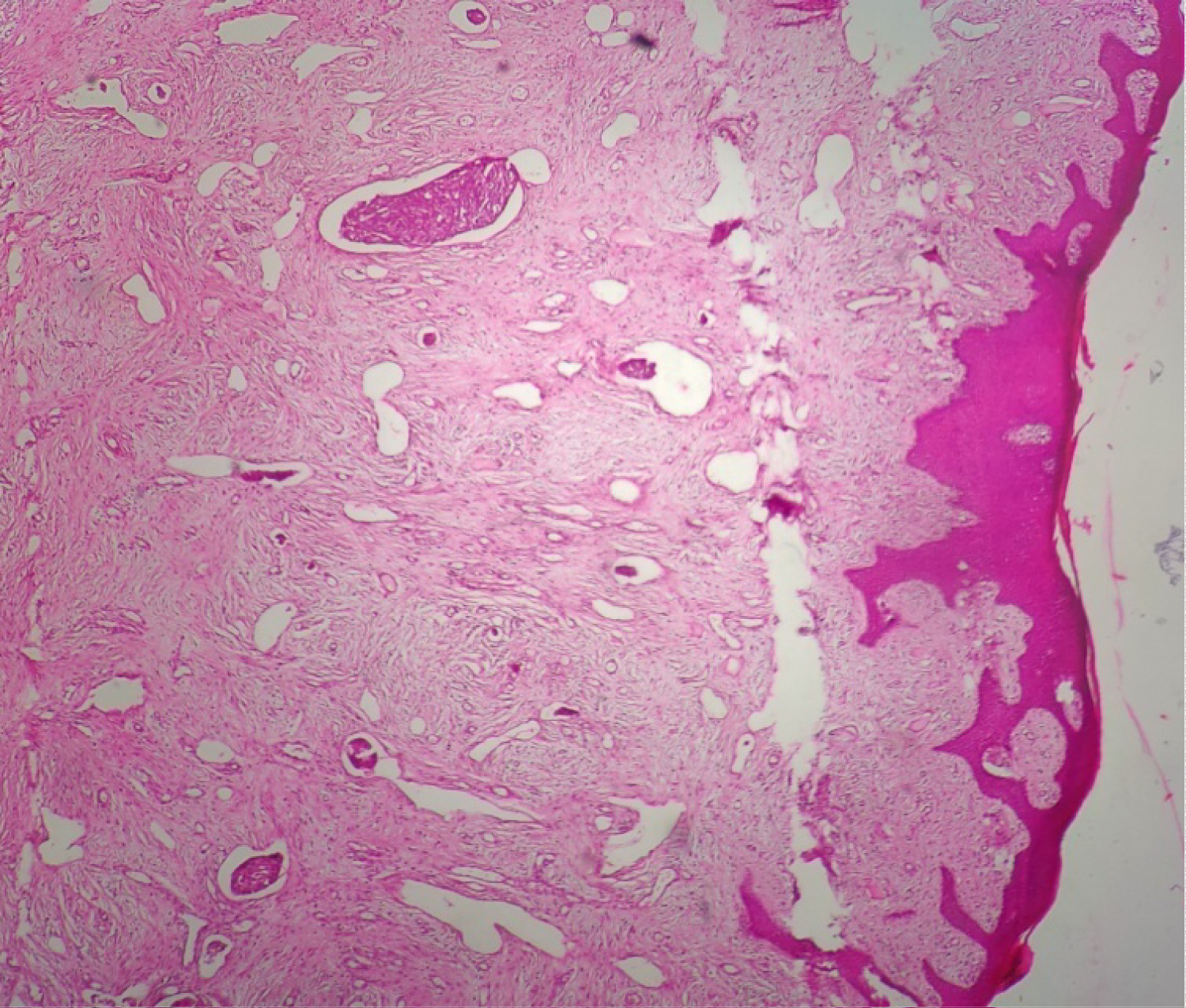 Figure 2: HEx10 showing intravascular tumor in the lamina propria.
View Figure 2
Figure 2: HEx10 showing intravascular tumor in the lamina propria.
View Figure 2
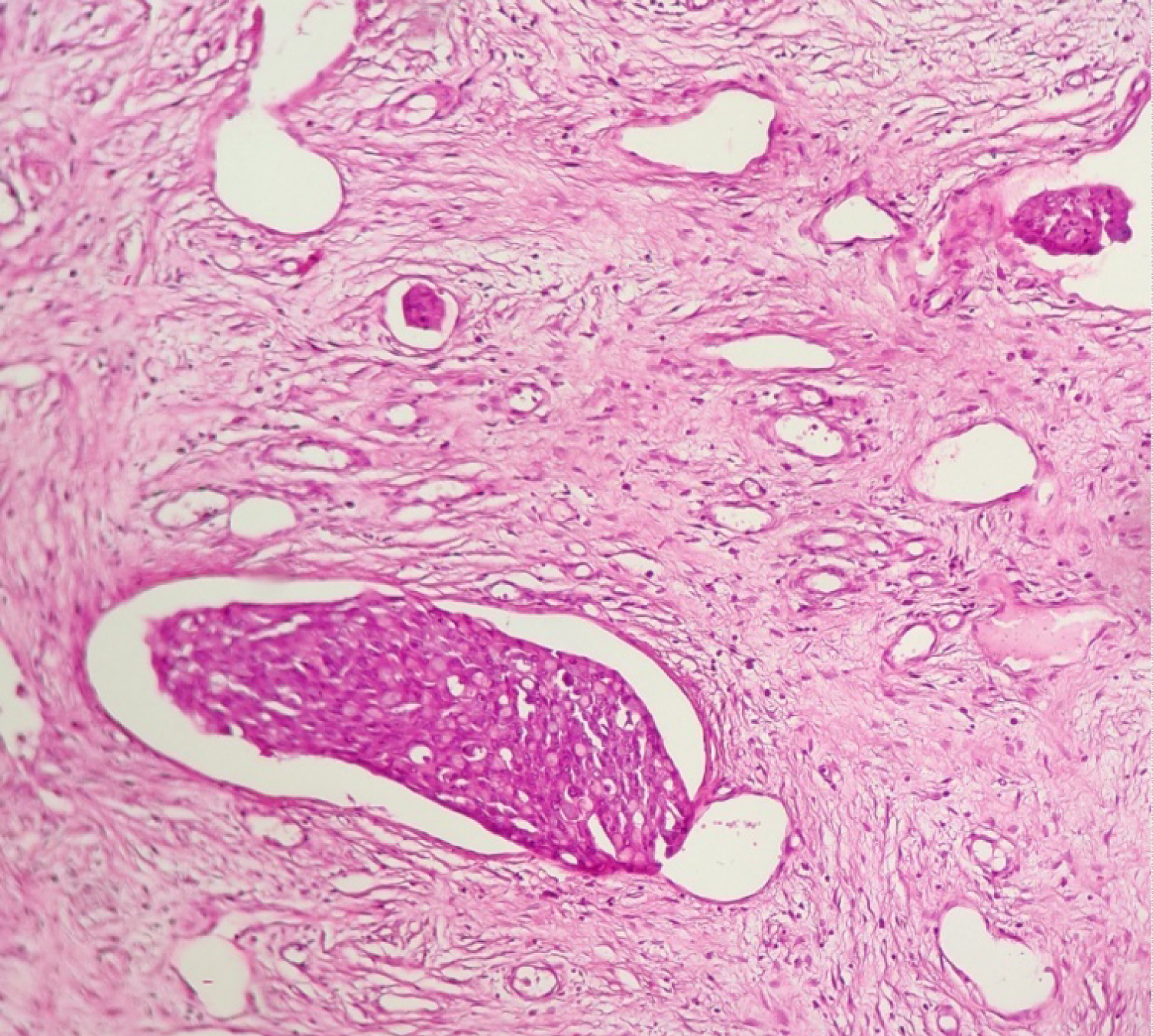 Figure 3: HEx25 showing signes rings cells in the lumina of vessels.
View Figure 3
Figure 3: HEx25 showing signes rings cells in the lumina of vessels.
View Figure 3
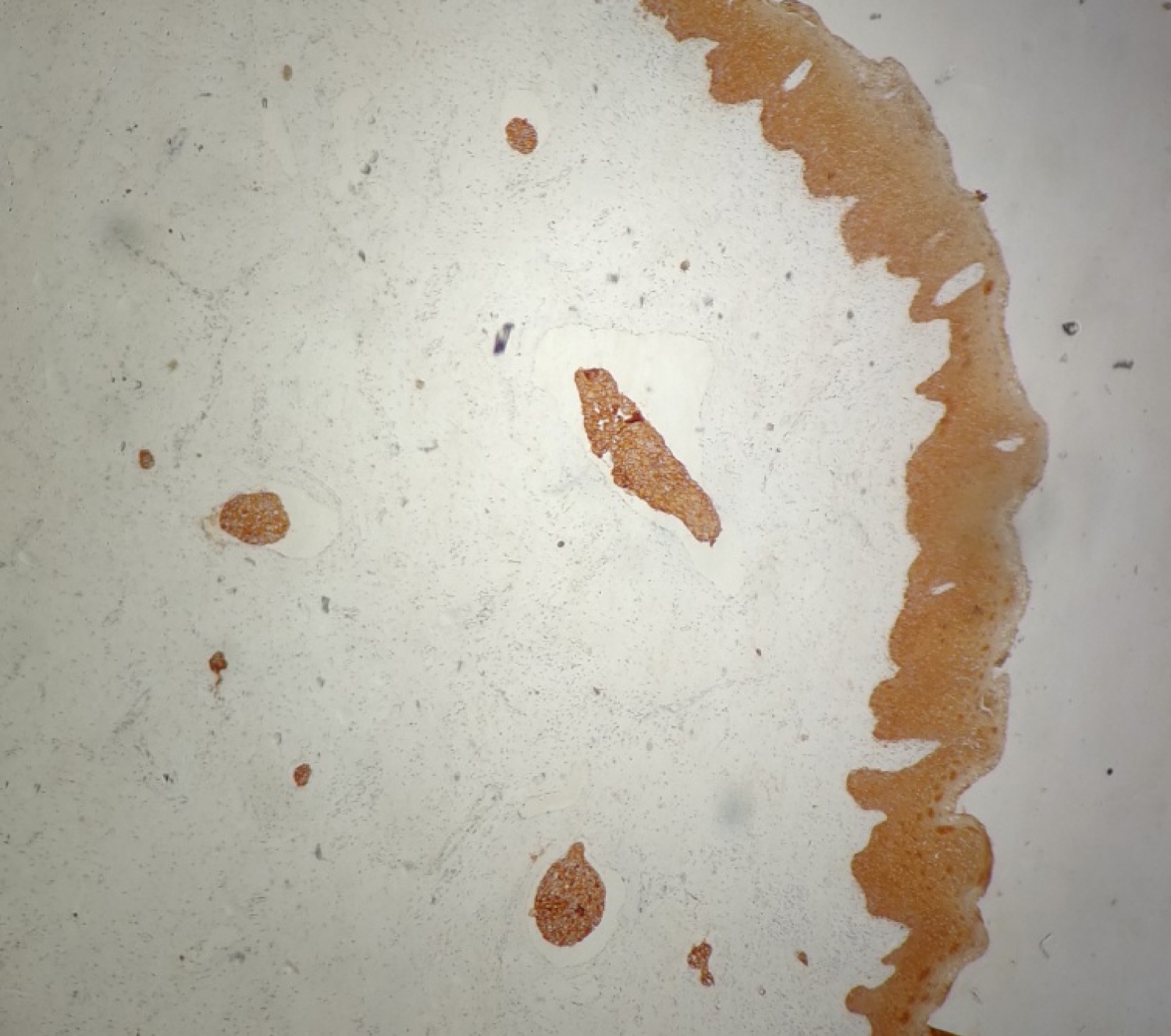 Figure 4: IHCx10 tumor cells are strongly positive for AE1/AE3.
View Figure 4
Figure 4: IHCx10 tumor cells are strongly positive for AE1/AE3.
View Figure 4
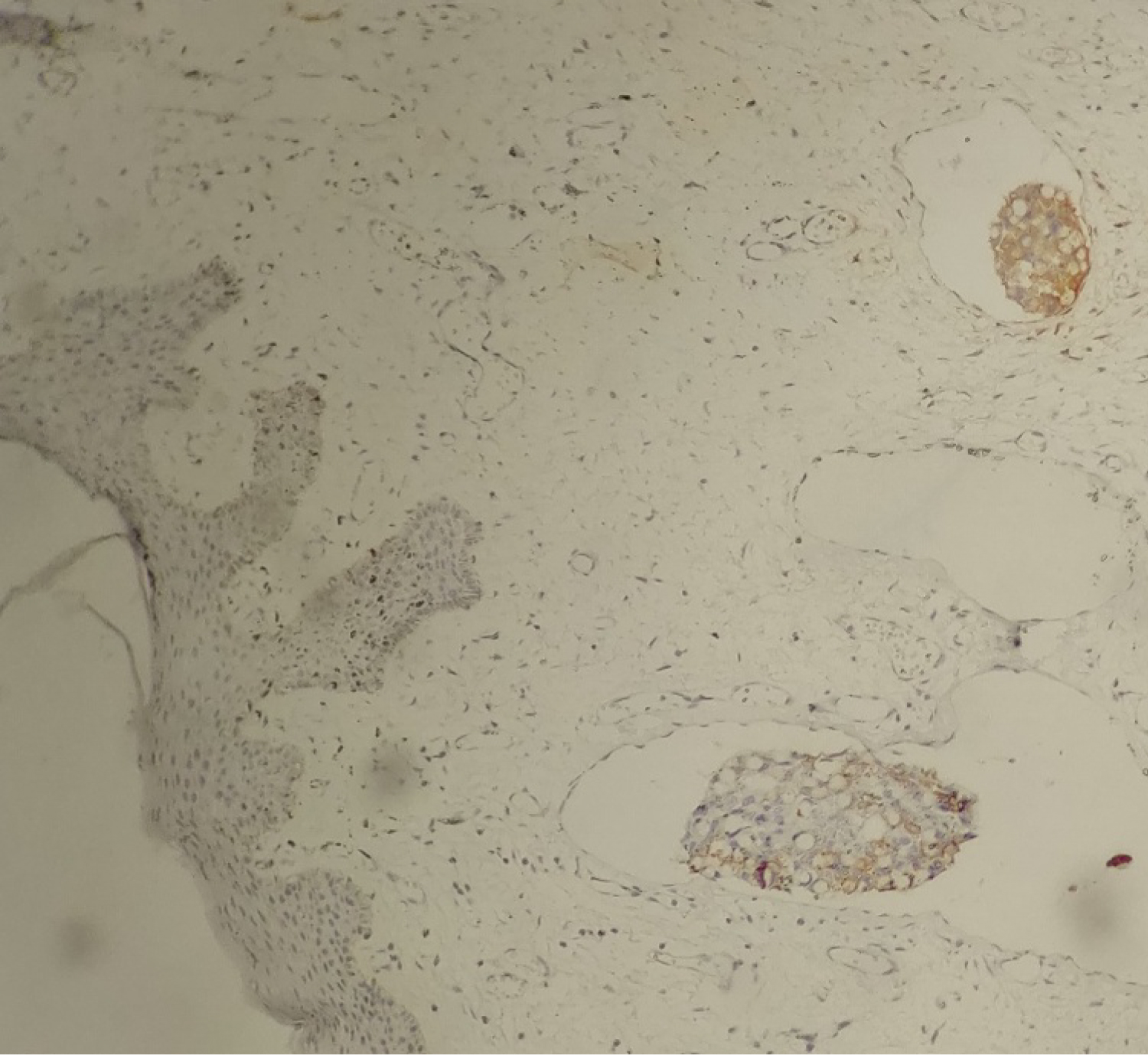 Figure 5: IHCx25 tumors cells are CK7 positive.
View Figure 5
Figure 5: IHCx25 tumors cells are CK7 positive.
View Figure 5
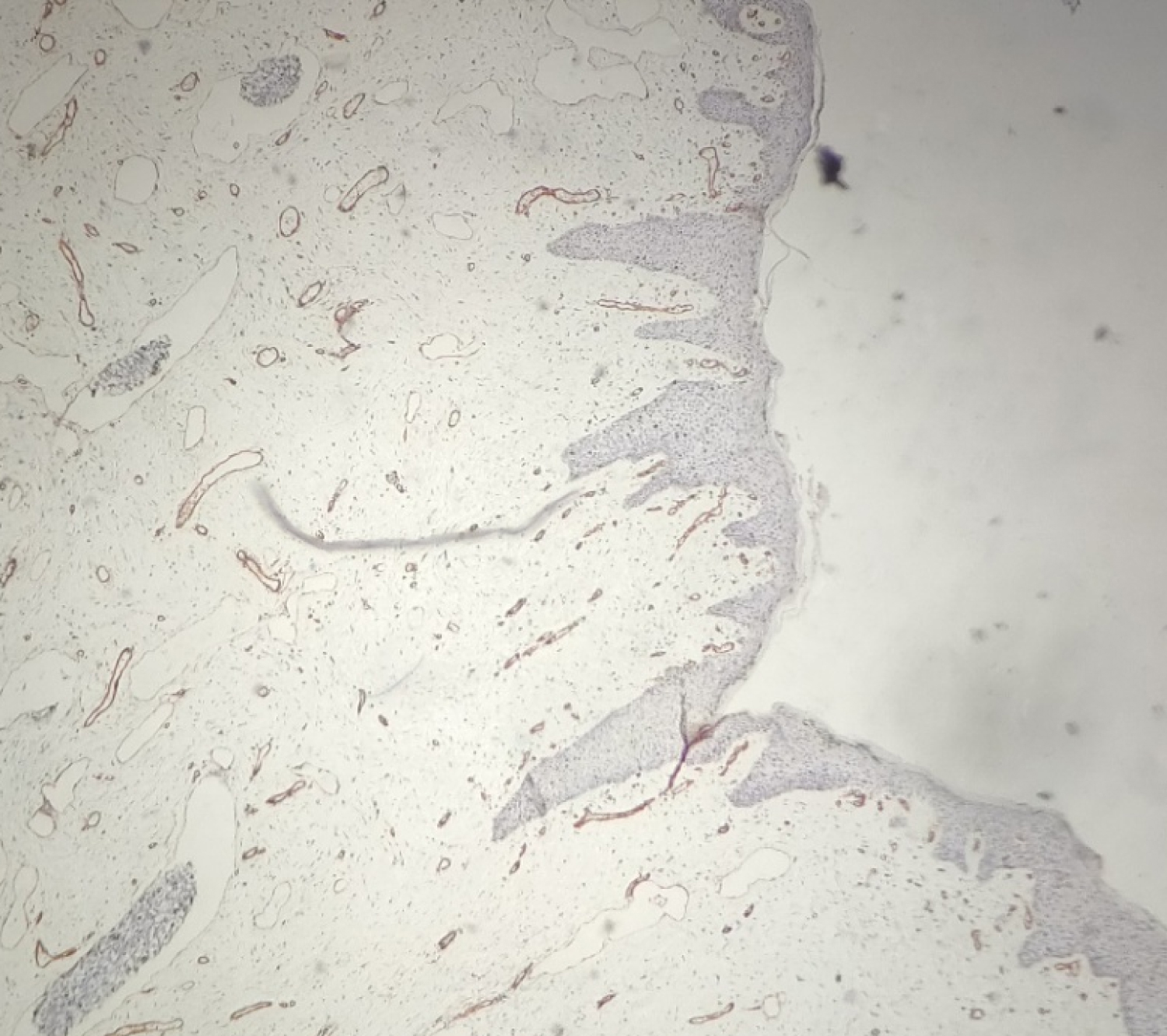 Figure 6: IHCx10 showing CD34 positive on the vessels.
View Figure 6
Figure 6: IHCx10 showing CD34 positive on the vessels.
View Figure 6
The diagnosis of a carcinomatous lymphangitis was made and the surgeon was informed of the diagnosis, which led to a more thorough examination which revealed that the patient was complaining of chronic epigastric pains resistant to conventional treatment. An endoscopy with gastric biopsy was made and returned in favor of a signet ring cell gastric adenocarcinoma.
After carrying out an extension assessment the computed tomography revealed nodules of peritoneal carcinosis, and the magnetic resonance imaging showed bilateral ovarian metastases.
The patient was put on palliative chemotherapy with a cure every 2 weeks. 3 months later the patient presented a severe cough, a chest radiograph revealed bilateral hilar lymphadenopathy. The patient died 12 months later.
The entire female genital tract is at risk for metastases occurrence from intragenital and extragenital primaries most commonly gastrointestinal adenocarcinomas [5]. The vulva is the least metastatic site of the female genital tract with the labia majora being the most common location [6]. It is noteworthy to mention that the occurrence of vulvar metastasis represents an end stage disease as it underlines, in general, a widespread incurable disease [2].
VLC is an invasion by tumor cells of the lymphatic vessels of the chorion. Clinically it can mimics benign lesion as angiomyofibroblastoma, cellular angiofibroma or hidradenoma papilliferum.
Neto, et al. reported the most common clinical presentation of vulvar metastasis occured in postmenopausal women (mean 54.8 years old) especially in labium majus mass [2]. Our case was also in labium majus but in young woman.
In both series reported by Neto and Mazur, malignant tumors that frequently give vulvar metastasis are from lower gastrointestinal tracts including colorectum and appendixsis. Our case was not consistent with this statistical finding and shows a gastric origin. Other primary malignancies in the breast, lung, appendix, and urinary system, as well as even lymphoma, have been reported [2,5].
VLC is not commonly observed. Despite its very low incidence, a precise etiological diagnosis of metastases is essential because the treatment and the prognosis are different. Therefore, metastases should also be suspected in patients with vulvar masses.
The authors declare that they have no competing interests.