The hemoglobin inherited disorders are one of the commonest single-gene disorders in man. Frequency of carriers of hemoglobinopathies varies from 3 to 17% in India. It is estimated that around 5.35% in India are the trait of Sickle cell, hemoglobin D, and hemoglobin E. However, the definite identification of disorders of hemoglobin synthesis can be achieved only by DNA analysis. In Indian scenario, family studies can be equally efficacious and cost-effective tool on HPLC and might be useful. This is a prospective study of 28 months. Total 37 families comprising of 114 family members were studied. Patients attending OPD and indoor admissions of institute with anemia, hepatosplenomegaly, showing sickling test and/or fetal fraction positive were included in the study along with their family members.
Our study reported 11 cases of β-thalassemia major, 16 cases of Sickle- β-thalassemia, 7 cases each of sickle cell disease and Hb-E trait, 4 cases of Hb-E β-thalassemia, 2 cases of Hb-Lepore trait along with 41 cases of β-thalassemia trait, 20 cases of Sickle cell trait and 1 case of β-thalassemia intermedia from the study population.
To detect "asymptomatic traits", for early diagnosis of variants presenting late in life, for accurate diagnosis of complicated heterozygotes in absence of molecular studies and prevention of propagation of hemoglobinopathies in families by prudent premarital and prenatal studies, family studies are indispensable. Community based targeted family studies along with HPLC can be an effective tool in reduction of disease burden.
Hemoglobinopathies, Family study, HPLC
SCA: Sickle Cell Anemia; HbS: Hemoglobin S; SCD: Sickle Cell Disease; SS: Sickle Homozygous; AS: Sickle Heterozygous; Sβ thal: Sickle Beta Thalassemia; HPLC: High Performance Liquid Chromatography; SC: Scheduled Caste; ST: Scheduled Tribes; HbA: Adult Hemoglobin; HbF: Fetal Hemoglobin; ISC: Irreversible Sickle Cells; VOC: Vaso-occlusive Crisis; ACS: Acute Chest Syndrome; BMA: Bone Marrow Aspiration; fl: Femtolitres; Hb: Hemoglobin; HbA2: Delta Hemoglobin; HCT: Hematocrit; RDW: Red Cell Distribution Width; Retic: Reticulocyte
The inherited disorders of blood include haemoglobinopathies together of the main public health problems in India. It has been estimated that there would be about 45 million carriers and about 15,000 infants born each year with haemoglobinopathies in India. The carrier frequency of haemoglobinopathy varies from 3 to 17% in several population groups of India. Thus, there is an incredible amount of burden of haemoglobinopathies in India. The prevalence of sickle cell gene is found to be 0 to 18% in North East India, 0 to 33.5% in Western India, 22.5 to 44% in Central India and 1 to 40% in Southern India. Gene frequency of Hbs varies between 0.031 to 0.41%, wide variability in the prevalence of Hbs trait is observed in population groups within small geographical areas [1,2].
The world health organization (WHO) recognizes thalassemia as the most prevalent genetic blood disorder in the world, found in more than 60 countries with a carrier population of 150 million (Cao and Galanelio), approximately 250 million people are heterozygous for beta thalassemia and at least 2,000,000 affected homozygotes are born annually, while some other reports have shown 3% to 10% of the world's population carries a thalassemia gene. The highest frequency of beta thalassemia trait is reported in Gujrat followed by Sindh, Punjab, Tamil Nadu, South India, and Maharashtra [3].
Automated cation-exchange High Performance Liquid Chromatography (HPLC) has emerged as an excellent screening tool for diagnosing these abnormal hemoglobin's/thalassemic states. The diagnosis of hemoglobin synthesis disorders can be seen ideally only by DNA analysis but, in Indian scenario with paucity of funds and resources, family studies on HPLC could be alternative tool. Family study is an equally efficacious and cost-effective tool [4].
The present study is useful to generate detailed data of role of family study in hemoglobin disorders. So that health care resources are often successfully targeted at them. Carrier identification in family would be useful to create awareness and offer them like partner selection, prenatal diagnosis, and contraception.
After the approval of the college and hospital ethical committee, the study of Hemoglobin disorders including double heterozygosity by High Performance Liquid Chromatography (HPLC) was carried out in Department of Pathology of a tertiary care hospital from July 2011 to November 2013 (2 years and 4 months).
All Patients ready to give written informed consent and attending OPD/admitted in IPD having anemia, hepatosplenomegaly and positive sickling test and/or fetal fraction test were considered as target population. Their family members were subjected for HPLC.
With all aseptic precautions, 4 ml of venous blood was withdrawn from patients. 2 ml blood was collected in 4% K2 EDTA (Ethylene diamine tetra-acetic acid) anticoagulant bulb and from remaining 2 ml; serum was separated for further processing. Followings parameters were analyzed.
Haematological parameters: Hb, Hct, MCV, MCH, MCHC, and RDW were obtained using Cell-Tech is an automatic multi-parameter blood cell counter. Sickling test, Solubility test, Fetal fraction and reticulocyte count were performed. Peripheral blood smear examination was done to evaluate red cell morphology.
Standard method: Primary screening for hemoglobinopathy was done with the help of BIO-RAD VARIANTS [5], β-thalassemia short program cation-exchange High Performance Liquid Chromatography, which works on the principle of separating the positively charged haemoglobin by adsorbing them to a negatively charged stationary phase and eluting them with a mobile phase at a rate related to their affinity for the stationary phase. Interpretation of the migration pattern of the test samples was undertaken by comparing them to those of known adult as well as fetal controls.
Clinical findings were correlated with radiological investigations like USG abdomen, X-ray chest and X-ray hip joints were done and other specific investigations as and when required. Hematological parameters and HPLC pattern were analyzed and conclusive diagnosis could be given in most of the cases. Intermediate cases were further investigated with the study of family members accompanying the cases to confirm the diagnosis and to determine ethnic background.
In the present study 1260 patients were screened who were suspected cases of hemolytic anemia, out of which 600 (47.65%) patients were having hemoglobinopathy and 660 (52.38%) were normal. Amongst them we have selected 37 families for study of hemoglobinopathies which comprises of total 114 family members. Out of 114 members 111 members were found to have hemoglobinopathies and 3 of them were normal.
The cases are grouped according to complete hematological workup, HPLC study and family study cases are classified as: Group-A Sickle Cell Trait (SCT), Group-B Sickle Cell Diseases (SCD), Group-C Beta Thalassemia Trait (BTT), Group-D Beta Thalassemia Major (BTM), Group-E Beta Thalassemia Intermedia (BTI), Group-F Sickle Cell - Beta Thalassemia (SBT), Group-GHbE Trait, Group-HHbE - Beta Thalassemia, Group-IHbLepore Trait (Table 1).
Table 1: Cases are grouped as follows with the percentage (N = 111). View Table 1
Patients whose blood sample was not accompanied by family members' blood sample were not included in the study. Family member's blood sample was not accompanied at the department due to distance, remarriage, death, and poor socioeconomic status, reluctance in father, lack of awareness, misconceptions, alcoholic father and psychological reasons.
Maximum cases (41) of group C (β thalassemia's trait) of which 19 cases are seen in 21-30 years of age, single case each seen in 51-70 years of age, followed by group A (sickle cell trait) 22 cases are seen in which more cases in 21-30 years of age, and group F (sickle β thal) in which 16 cases of which 6 cases are seen in 11-20 years of age.
In Group-A i.e. sickle cell trait No. of male patient are 12 and female patient 8 with M: F ratio 1.5:1, followed by in Group-C i.e. Beta Thal Trait No. of male patient 21 and female patient 20 with M: F ratio 1.05:1, in Group-F i.e. Sickle Beta Thal No. of male patient 8 and female patient 8 with M: F ratio 1:1.
In the study out of 37 family's history of consanguinity was present in 11 (29.72%) families. Caste wise distribution of families, Buddha (44%) was the most common ethnic background among all the groups followed by Muslims (17%). In HbE-β thalassemia cases, most common ethnic background was Buddha followed by Muslim.
Majority of the cases presented clinically with Pallor i.e 33 cases (41%) next common presentation was abdominal pain. Other clinical features were jaundice, hemolytic facies, and abdominal pain (Figure 1).
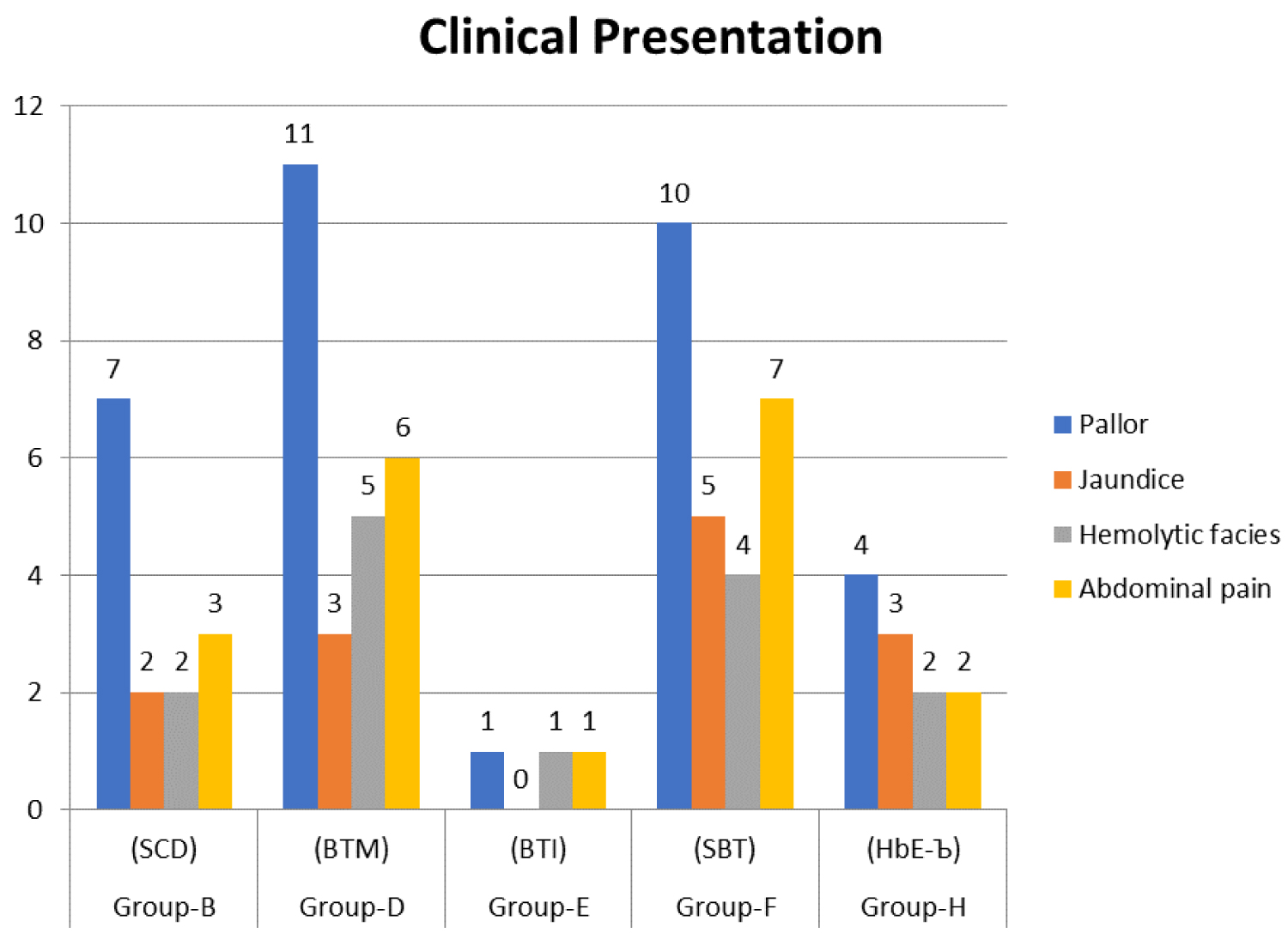 Figure 1: Bar Diagram: Showing clinical presentation of index cases among all groups.
View Figure 1
Figure 1: Bar Diagram: Showing clinical presentation of index cases among all groups.
View Figure 1
Out of 37 cases 19 cases presented with Splenomegaly. Out of 19 cases 10 cases were found in 0-10 years age group followed by 7 cases in 11-20 years age group. So, the maximum cases of Splenomegaly were found in first decade.
Maximum cases of hepatomegaly were found in first decade of life (66%) of which maximum No. of patients with hepatomegaly were found in Group-D (BTM).
Lowest Hb was found in Group D i.e. Beta thalassemia major and highest level of Hb was found in Group F i.e. Sickle Beta thal. Lowest RBC count was found in Group D i.e. Beta thalassemia major and highest RBC count was found in Group F i.e. Sickle Beta thalassemia. Lowest MCV was found in Group D i.e. Beta thalassemia major and highest MCV was found in Group B i.e. Sickle cell disease. Lowest MCH was found in Group H i.e. HbE-Beta thal and highest MCH was found in Group B i.e. Sickle cell disease.
In the present study of total 114 members of 37 families, the average age of presentation of sickle cell disease was 11.28 years, β-thal major was 2.31 years, sickle β-thal was 19.12 years and Hb-E β-thal was 24.5 years. Similar findings were observed by R.S Balgir and Tyagi, et al. [6,7].
Out of 7 cases of sickle cell disease, 6 were male and 1 was female, 11 cases of β-thal major, 8 were male and 3 were female, 16 cases of sickle β thal 8 were male and 8 were female and 4 cases of Hb E β thal 3 were male and 1 was female. Similar observation was made by R.S Balgir & Tyagi, et al. [6,7].
Pallor is the most common clinical presentation observed in sickle cell disease patients (100%), Beta thal major (100%) and HbE Beta thal (100%) in this study. Abdominal pain was the next most common presentation in sickle cell disease (42.85%), Beta thal major (54.54%), Sickle beta thal (43.75%), HbE beta thal (50%), similar findings were seen by R.S Balgir & Tyagi, et al. [6,7], except they observed vaso-occlusive crisi 31% and 81.3% respectively.
In the present study the most commonly sickle cell disease affected ethnic group is Buddha followed by banjara and muslim. Most common Beta thalassemia affected ethnic group is Buddha followed by muslim. Most common Sickle beta thalassemia affected ethinic group is Buddha followed by muslim and banjara. Most common Hb-E Beta thalassemia affected ethnic group is Buddha followed by muslim. Kate SL reported high prevalence amongst SC, ST and OBC in State of Maharashtra. Kamble M, Chaturvedi P observed maximum incidence of sickle hemoglobinopathy in Mahars (70%) followed by Kunbhis (8%) and Telis (6%) [8,9]. Bhatia and Rao found prevalence of sickle cell disorder is very high amongst tribal and scheduled caste population group where carrier frequencies range between 5-40% or more. Dangi, et al., found Muslims, Sindhis and Tribal population the major carriers of sickle gene [10,11].
Patel DK had quoted that, in India the practice of endogamy and consanguineous marriages are very common, and since β-thalassemia and hemoglobin S (HbS) are relatively frequent in India, compound heterozygosity for these two disorders (βS/ β-thalassemia), usually expressed in a severe type of disease, is not rare [12].
Tariq HA narrated that, the main factor, which may increase or decrease the risk of having an affected child, is the parents' relationship. The risk of hemoglobinopathies increases with consanguineous marriages. This is because all the hemoglobinopathies are transmitted only through heredity [13]. In the present study there were 11 families out of 37 (29.72%) showing history of consanguinity. Consanguinity is commonly practiced in Buddha and Muslim communities.
In the present study splenomegaly was found in 2 (28%) patients of sickle cell disease. In the Beta thalassemia major patient's splenomegaly was found in 5 (45.45%) patients. In the Sickle beta thalassemia patient's splenomegaly was found in 8 (50%) patients. In the HbE beta thalassemia patient's splenomegaly was found in 3 (75%) patients. Tyagi, et al. in their study on sickle cell syndrome, found that pallor (62.5%), jaundice (75%), painful crisis (62.5%) and leg ulceration (25%) were more frequent in sickle cell disease than in double heterozygous Sβ Thal patients [7]. They also found that the frequency of splenomegaly was significantly higher (75%) in patients with Sβ Thal than Sickle cell disease. Panigrahi I, et al. in their study on HbE β Thal patients found pallor as the commonest feature (100%), splenomegaly in 74% cases, hepatomegaly in 65% cases, jaundice in 57% cases [14]. Shah SJ, et al. reported - 35 out of 35 cases with pallor (100%), 31 cases with splenomegaly (88.6%), 25 cases with hepatomegaly (71.4%) and 8 cases with icterus (22.9%) [15]. RS Balgir, et al. found splenomegaly in 17.1% of cases. Splenomegaly is higher in patients with Sβ thalassemia than SS disease [6]. Patel DK stated that persistent gross splenic enlargement is peculiarity of Indian sickle disorder patients [12].
Present study means hematological parameters in all Age groups: In the study of various authors, it is observed that low hemoglobin levels in thalassemia major are < 7 g%. Similar was the finding in present study with thalassemia major group showing low hemoglobin levels (mean 4.5 g%) as compared to other groups. Similarly, hemoglobin levels in sickle beta thalassemia were < 10.1 g%. In the present study mean hemoglobin levels in sickle beta thalassemia were 6.96 g%. Hemoglobin levels in Hb-E beta thalassemia were found to be < 6.56 g%. In the present study the mean hemoglobin level in Hb-E beta thalassemia was found to be 5.5 g% [7,14-17].
In the present study the HbF level in Beta thalassemia major was high > 90% as compared to the other studies in which it was quite variable < 90%. This may be due to the reason that the cases included in the present study did not receive any blood transfusion [17-21].
In the study of Ambekar SS, Rao S, et al. C Vani and S Mamta and in the present study the Hb S levels were > 60% (except in Dangi, et al. study). In all the above studies the average Hb F levels were high > 15%, in the present study it was 16.99%. The average HbA2 level in present study was 5.48% [11,19,20,22].
In all the studies Fucharoen S, et al. Sachdev R, Rao S, C Vani, et al. Jha BM, et al. in Hb-E beta thal patients, average HbF levels were above 15%. In the present study HbF level was (15.35). Average HbE variant levels were > 40% in all the above studies, in the present study it was 43.97% [11,14,18,22,23].
Our findings thus support that HPLC family studies is cheap, easily accessible, and equally efficacious compared to genetic studies. Family studies also help in premarital counselling and hence offer chances of like partner selection.
We are thankful to all technical staff of Department of Pathology, Govt. Medical College, Aurangabad (Maharashtra INDIA) for their help and support.
We sincerely acknowledge our institute colleague Dr. Mirza Shiraz Baig, Associate Professor in Pharmacology for his sincere help and assistance in medical writing of this article.
Nil.
Nil.
Facilities available in department of Pathology.
All authors have equally contributed during study and in drafting the article.