Extrarenal rhabdoid tumours appearing in the soft tissues are very rare and might be confused with other high-grade sarcomas harbouring rhabdoid features. We present a case and highlight the differential diagnosis, the ancillary techniques necessary to define its true rhabdoid nature, and the existence of hereditary syndromes.
Our case concerns a new-born boy with a congenital and rapidly growing mass in the soft tissue of the left thoracic wall, expanding from a few cm in diameter at the time of birth, to a mass measuring more than 20 cm after one month. Imaging based on US and MRI revealed a large soft tissue tumour invading the pleura of the left lung (Figure 1). Additional PET/CT demonstrated spread of the disease in the spleen, lymph nodes, muscle, cerebellum, skin and bone.
 Figure 1: Beelden case report rhabdoid tumor.
Figure 1: Beelden case report rhabdoid tumor.
Figure 1A: Ultrasound images with a high-frequency (12 MHz) linear probe from the lateral chest wall. The rib cage is demonstrated by the stars in (a) and (b). Intercostal protrusion of the mass is shown by the white arrow in (a), more posteriorly compared to (b). The larger component of the mass is located more anteriorly and consists of a large soft tissue component containing several areas of liquefaction/necrosis (white circles in (b)).
Figure 1B: Computed tomography (CT) images of the chest (total dose of the CT examination 3 mGycm) demonstrates a large mass in the left chest wall. Mild bulging of the left ribs medially is noted. The intercostal extension of the mass is demonstrated is seen with by the white arrows in (a). Heterogenous contrast enhancement is seen in (b) with nodular areas of enhancement (dashed white arrow in (a) and (b)) and necrotic areas (black arrow in (a) and (b)).
Figure 1C: Computed tomography (CT) images of the chest (total dose of the CT examination 3 mGycm).
Figure 1D: Magnetic resonance (MR) images with a T1 gradient echo image with fat saturation (a), diffusion-weighted image (DWI) with a b-value of 1000 and a T2 weighted turbo-spin echo image, all in the axial plane. The central necrotic areas are demonstrated in all three images by the white circle. The enhancing soft tissue component (white arrow in (a)) is also visible on DWI as bright signal (white arrow) and on T2 as mild hyperintense signal (white arrow). Furthermore, the intercostal extension of the mass within the chest on the lateral left side of the vertebral body is best seen on DWI (grey arrow in (b)).
View Figure 1
Needle biopsy revealed a solid tumour with collagenous to myxoid background, sheets and nests of pleiomorphic epithelioid cells with high mitotic activity. The nuclei were irregular and vesicular. The cytoplasm varied from clear to densely eosinophilic (Figure 2a). The initial histological differential diagnosis concerned Ewing sarcoma, congenital fibrosarcoma, pleuropulmonary blastoma, synovial sarcoma and rhabdoid tumour of soft tissue. Immunohistochemistry for CD 99, alfa smooth muscle actin and p53 was positive, there was faint and variable expression of keratins and s100. Desmin, myogenin, TdT and synaptophysin were negative. INI1 staining was negative as well (Figure 2b).
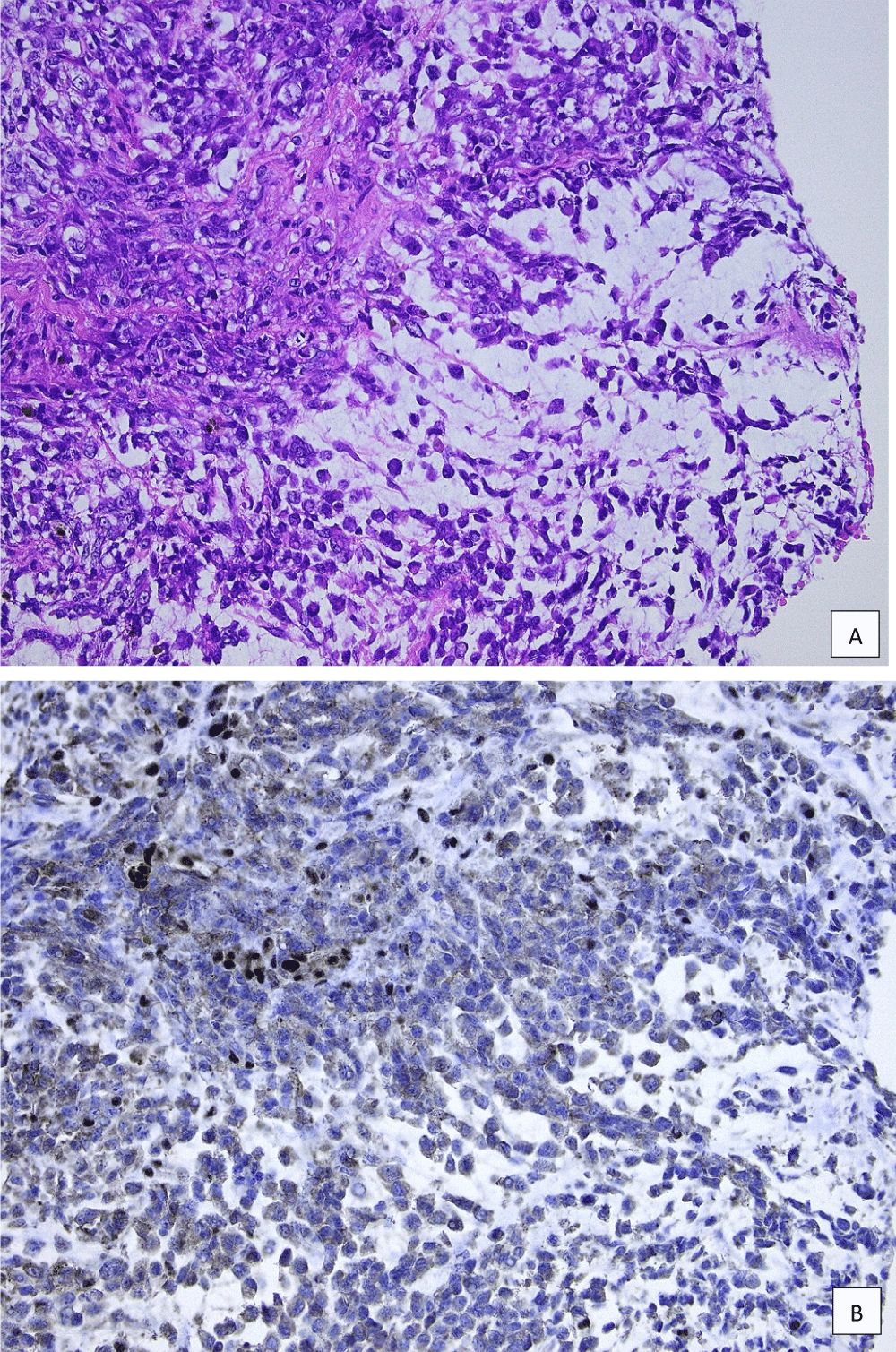 Figure 2: HE INI1 case report.
Figure 2: HE INI1 case report.
a) HE slide representative of the tumour; b) Immunohistochemical stain showing loss of INI1 expression, with some lymphocytes and endothelial cells serving as positive internal controls.
View Figure 2
Fluorescent in situ hybridisation on formalin fixed paraffin embedded material of the tumour showed no evidence for EWSR1 rearrangement (to exclude Ewing sarcoma). There was no loss of SMARCB1/INI1 in the PDGFB region (22q11) and no ETV6-NTRK3 rearrangement was found (excluding congenital fibrosarcoma). Eventually Next Generation Sequencing on the FFPE material showed a pathogenic nonsense mutation c.842G > A (p.Trp281*) in SMARCB1, ascertaining the diagnosis of a malignant rhabdoid tumour. Additionally, a heterozygous c.842G > A (p.Trp281*) nonsense mutation, causing a premature stop codon, was found in a blood sample of the patient. This mutation is seen in Rhabdoid Tumour Predisposition syndrome (RTPS). Three months after birth, palliative support was initiated because of progressive disease despite chemotherapy.
Malignant rhabdoid tumour is best known in the kidney and the central nervous system but primary lesions may appear in other organs, skin and soft tissue [1,2]. Extrarenal rhabdoid tumours of soft tissue are highly aggressive and seem to be confined almost exclusively to children. Congenital cases are reported in the literature and rhabdoid tumour predisposition syndromes 1 and 2 (RTPDS 1 and 2) have been described [3-5]. Most often extrarenal rhabdoid tumours of soft tissue present in a deep axial location, the neck or paraspinal [1].
Mutations causing inactivation or loss of the SMARCB1 gene (also known as INI1) on chromosome 22, deletions or translocations involving 22q11.2 and subsequent loss of INI1 expression by immunohistochemistry are essential for the diagnosis. These alterations are found in more than 90% of extrarenal rhabdoid tumours and in rhabdoid tumours of the kidney and central nervous system [1].
They are usually not found in other sarcomas harbouring areas of rhabdoid dedifferentiation except for epithelioid sarcomas [6]. These lesions may show (predominant) rhabdoid morphology on histological examination, show expression of vimentin and keratins (e.g. EMA) and distinction between epithelioid sarcoma and extrarenal rhabdoid tumour can be difficult, if not impossible. CD34 is expressed in about 40% of epithelioid sarcomas and should not be a feature of rhabdoid tumours. Epithelioid sarcoma appears more often in older patients and thus the age of the patient could be the only discerning factor.
Patients with malignant rhabdoid tumour of soft tissue present at a young age, often with disseminated disease and the aggressive course of the disease is most often fatal [1]. Patients presenting with multiple lesions, congenital disease or in the first years of life might suffer from hereditary syndromes RTPDS1 or 2. It is suspected that up to 35% of malignant rhabdoid tumours are caused by a germline mutation. RTPDS1 is an autosomal dominant condition caused by mutations in SMARCB1 (22q11.23), leading to malignant renal and extrarenal rhabdoid tumours and tumours of the central nervous system (e.g. hereditary schwannomatosis). RTPDS2 is caused by mutations in the SMARCA4 gene (located at 19p13.2), leading to malignant rhabdoid tumours and lesions in the ovary (e.g. small cell carcinoma of the ovary hypercalcaemic type). Our patient presented with a congenital rhabdoid tumour of soft tissue, disseminated disease at the time of presentation and rapid progression. The presence of a heterozygous c.842G > A(p.Trp281*) nonsense mutation in the SMARCB1 gene confirmed the presence of RTPDS1.
H Plasschaert collected the data, reviewed the literature and wrote the case report. F Deman collected the data and reviewed the manuscript. I Vanden Bempt and V Labarque reviewed the manuscript. M Aertsen supplied the radiology images and H Brems the genetics data, both reviewed the manuscript. R Sciot diagnosed the case, reviewed and revised the manuscript.