Solitary fibrous tumor is a rare soft-tissue neoplasm that is capable of metastasis of fibroblastic/myofibroblastic cell origin. Although most of the cases are pleural in origin, extrapleural locations are reported with increasing frequency. Extrapleural cases are mostly seen in retroperitoneal location. Malignant forms are much rarer.
A 61-year-old male patient was admitted with complaints of dysuria, low back pain and constipation. Radiological investigation revealed a retroperitoneal mass lesion. The excised mass was macroscopically well-circumscribed without capsule and was measured as 12 × 10 × 8 cm. The tumor was hypercellular and it contained necrosis areas. Peripheral soft tissue invasion was noted in the tumor. Tumor showed a vascular pattern that reminiscent of a hemangiopericytomatous pattern. Tumor were positive with CD34, bcl-2, EMA, vimentin, PAX8 and STAT6 in immunohistochemical analysis. The case was diagnosed as retroperitoneal solitary fibrous tumor. The tumor was compatible with the malignant category due to widespread necrosis, increased cellularity and high mitotic index.
A 61-year-old male patient was admitted with complaints of dysuria, low back pain and constipation. Radiological investigation revealed a retroperitoneal mass lesion. The excised mass was macroscopically well-circumscribed without capsule and was measured as 12 × 10 × 8 cm. The tumor was hypercellular and it contained necrosis areas. Peripheral soft tissue invasion was noted in the tumor. Tumor showed a vascular pattern that reminiscent of a hemangiopericytomatous pattern. Tumor were positive with CD34, bcl-2, EMA, vimentin, PAX8 and STAT6 in immunohistochemical analysis. The case was diagnosed as retroperitoneal solitary fibrous tumor. The tumor was compatible with the malignant category due to widespread necrosis, increased cellularity and high mitotic index.
In order to increase our knowledge about the clinicopathological features and biological nature of these tumors, there is a need more cases of extrathoracal/extrapleural solitary fibrous tumor.
Solitary fibrous tumor, Malignant features, Hemangiopericytoma, STAT6, Retroperitoneal location
SFT: Solitary Fibrous Tumor; SS: Synovial Sarcoma; GIST: Gastrointestinal Stromal Tumor; F-DFSP: Dermatofibrosarcoma Protuberans Showing Fibrosarcomatous Features; DLPS: Dedifferentiated Lposarcoma; HPF: High Power Field; WHO: World Health Organization; MRI: Magnetic Resonance Imaging
Solitary fibrous tumor (SFT) accounts for less than 2 percent of all soft tissue tumors [1]. Although the most common localization site is the pleura, the cases with extrapleural/extrathoracal location are increasingly being described [2]. Thirteen to fourteen percent of SFTs are extrapleural/extrathoracal. Localized cases of head, neck, intracranial, retroperitoneal regions, pelvis and genitourinary tract have been reported [3,4]. SFT cases with malignant histopathological features have also been reported in the literature.
Here, we present a case of SFT who presented with retroperitoneal malignant histopathological features in a 61-year-old man.
A 61-year-old male patient who presented with dysuria, low back pain and constipation complaints had narrowing of the rectum lumen by rectal digital examination. Colonoscopy showed an external compression to the rectum. Pelvic computed tomography showed a rectangular mass of 110 × 90 mm with a smooth contour in the right lateral side of the sigmoid colon, extending to the pelvis and compressing the prostate gland externally. Magnetic resonance imaging (MRI) revealed a mass of 126 × 119 × 99 mm T1 cross-sections with light hyperintense in the area corresponding to the prostate region, and a solid mass with vascular and fibrotic components showing significant heterogeneous hyperintensity in T2 sections (Figure 1). The totally resected mass was macroscopically well-circumscribed without capsule and was measured as 12 × 10 × 8 cm. The mass was multilobulated and the cross-section was in solid character and gray-white in color. Frequent bleeding and necrosis foci were present (Figure 2A). In the microscopic examination, the tumor was consisted of hypercellular multilobulated areas separated by edematous and hyalinized hypocellular bands. Ischemic necrosis and tumor necrosis areas were observed as large multifocal areas (Figure 2B). A focus of adjacent peripheral soft tissue invasion was noted in the periphery of the tumor (Figure 2C). Tumor stroma showed a vascular pattern in many areas and it was compatible with hemangiopericytoma in some areas while it was reminiscent a hemangiosarcoma in some areas (Figure 2D). Other areas showed vaguely vascular spaces consisting of crossing short bundles of tumor cells in hyalinized stromal areas (patternless pattern) (Figure 2E). Tumor cells had limited spindled cytoplasm and mild elongated plump nuclei with vesicular chromatin pattern (Figure 2F). Mitotic index was high in some areas [mitotic figure; 10/10 in a high power field (HPF)] (Figure 2G). Immunohistochemical analysis revealed a diffuse strong cytoplasmic staining with CD34 and bcl-2, multifocal cytoplasmic staining with EMA and vimentin, focal weakly membranous expression with CD99, diffuse nuclear expression with PAX-8 and STAT6 (Figure 2H, Figure 2I and Figure 2J). No expression was observed with C-kit, PDGFRA, DOG-1, SMA, MSA, desmin, factor-VIIIA, pan-cytokeratin, calretinin, CD10, PSA and S100. The Ki67 index was 10%. The case was evaluated as histopathologically compatible with solitary fibrous tumor with retroperitoneal location. The tumor was evaluated in the malignant category due to widespread necrosis, increased cellularity and high mitotic index. There were no signs of recurrence/metastasis in our case at the 12th postoperative month.
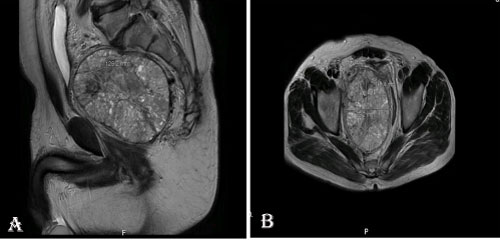 Figure 1: The images of vertical (A) and horizontal (B) plan of a well-circumscribed retroperitoneal solid mass lesion on the pelvic MRI of the patient. View Figure 1
Figure 1: The images of vertical (A) and horizontal (B) plan of a well-circumscribed retroperitoneal solid mass lesion on the pelvic MRI of the patient. View Figure 1
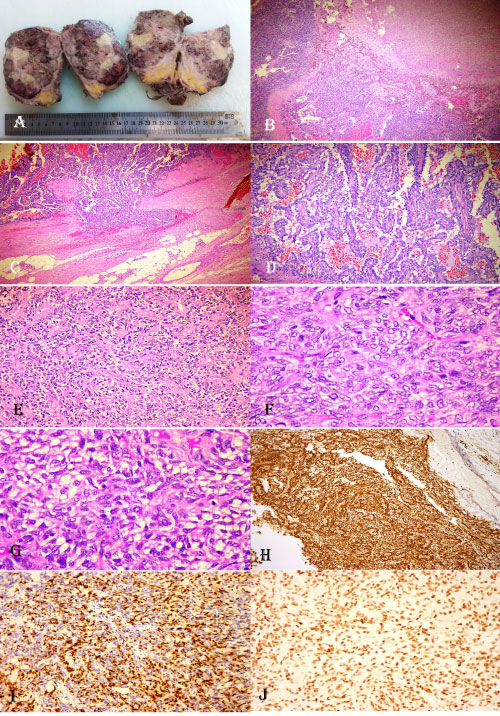 Figure 2: (A) Macroscopic appearance of the retroperitoneal pelvic mass lesion. Multiple foci of bleeding and necrosis on cross-sections of the multilobated and unencapsulated tumoral lesion with relatively well circumscribed margins; (B) An appearance of hemangiopericytomatous pattern adjacent to a large necrosis area at low power magnification (HE, X5); (C) Invasion area into neighboring soft tissues to the tumor (HE, X10); (D) A high power magnification of hemangiopericytomatous pattern (HE, X30); (E) A view from the area of patternless pattern in the tumor (HE, X30); (F) The high power appearance of tumor cells with oval, slightly fusiform-shaped and vesicular chromatin pattern (HE, X40); (G) A tumor area with increased mitotic figures (HE, X40); (H) Diffuse CD34 positivity in tumor (DAB, X20); (I) Nuclear PAX8 expression in tumor (DAB, X20); (J) Nuclear STAT6 expression in tumor (DAB, X20). View Figure 2
Figure 2: (A) Macroscopic appearance of the retroperitoneal pelvic mass lesion. Multiple foci of bleeding and necrosis on cross-sections of the multilobated and unencapsulated tumoral lesion with relatively well circumscribed margins; (B) An appearance of hemangiopericytomatous pattern adjacent to a large necrosis area at low power magnification (HE, X5); (C) Invasion area into neighboring soft tissues to the tumor (HE, X10); (D) A high power magnification of hemangiopericytomatous pattern (HE, X30); (E) A view from the area of patternless pattern in the tumor (HE, X30); (F) The high power appearance of tumor cells with oval, slightly fusiform-shaped and vesicular chromatin pattern (HE, X40); (G) A tumor area with increased mitotic figures (HE, X40); (H) Diffuse CD34 positivity in tumor (DAB, X20); (I) Nuclear PAX8 expression in tumor (DAB, X20); (J) Nuclear STAT6 expression in tumor (DAB, X20). View Figure 2
SFT is included in the category of tumors with fibroblastic/myofibroblastic origin with low metastasis capacity in the current World Health Organization (WHO) classification of soft tissue tumors [5,6]. The SFT was first described by Klemperer and Rabin in 1931 as a pleural-specific neoplasm [7]. Development of immunohistochemistry revealed that many cases of deep soft tissue hemangiopericytoma were actually SFT and many cases of SFT showed extrapleural/extrathoracal location [8]. Therefore, in the current WHO classification of soft tissue tumors, the term hemangiopericytoma was removed and mesenchymal neoplasms of similar morphology were defined as SFT [8]. SFTs do not show sex discrimination and they can be seen at any age, but they are more common in fifth to seventh decades [2,9,10]. Extrapulmonary/extrathoracal SFTs may occur as a painless mass lesion. Abdominal distension, abdominal pain and weight loss are the most common findings in intraabdominal SFTs. Small-sized tumors tend to be asymptomatic [2]. Our case's age was within the limits reported in the literature and our case had similar symptoms such as dysuria, low back pain and constipation. SFTs with intraabdominal/intrapelvic location may reach large sizes (> 20 cm). SFTs are well-demarcated masses with homogeneous cross-section. Radiologically, they show cystic areas, calcifications, and bleeding foci [11,12]. Radiological images of malignant forms may be in heterogeneous character because of macroscopically corresponding foci of necrosis and hemorrhage. In addition to this, infiltrative border features may be seen because of tumoral infiltration to neighboring soft tissues. However, our case' radiological images showed non-infiltrative, smooth border features. The definitive diagnosis of SFT is possible with histopathological examination. In addition to the hemangiopericytomatous pattern, SFT may be characterized by a "patternless pattern" which may be characterized by random sequencing of ovoid cells in hypo and hypercellular areas within hyalinized stromal areas [2]. The hemangiopericytomatous pattern is seen in many benign and malignant mesenchymal neoplasms including SFT. Many cases with extrapleural/extrathoracal locations that were previously diagnosed as hemangiopericytoma are accepted to be SFT by the advances of immunohistochemistry [8]. Therefore, immunohistochemistry is important in diagnosis. Expressions of vimentin, CD34, CD99 and bcl-2 are the characteristic immunophenotypes for SFT [2]. SFTs can be discriminated from other mesenchymal neoplasms with similar histomorphology and partially similar immunophenotype [gastrointestinal stromal tumors (GIST), smooth muscle tumors and nerve sheath tumors] thanks to they are negative for SMA, desmin, S100, c-kit and DOG-1 [2]. With the increasing of awareness for extrapleural/extrathoracic SFTs, malignant SFT cases have been also reported more frequently. Approximately 20% of SFT cases have aggressive clinical features [13]. Histomorphological clues of malignancy are hypercellularity, nuclear pleomorphism, necrosis/hemorrhage and increased mitosis. In addition, increased tumor size, infiltrative border characteristics and advanced age are also important [14,15]. The threshold value for tumor size was 10 cm according to Shoubbege, while this was 15 cm according to Demicco [14,15]. Increased mitosis (> 4/10HPF) and age over 55 year have been associated with increased malignancy risk [14,1]. Malignant histological features may not always be associated with aggressive clinical behavior. The opposite is also true. Fully benign SFTs have been reported to behave locally aggressive [4,16]. Nevertheless, SFTs with malignant histomorphology have been reported to have a higher rate of recurrence (8% versus 63%) and distant metastases [17]. Dedifferentiated SFT forms have been reported in addition to malignant SFTs [18]. Dedifferentiation refers to the sudden transition from a low-grade sarcoma area to a high-grade sarcoma area. Recognizing and not to skipping of this feature are important for predicting poor prognosis. A total of 23 dedifferentiated SFT cases have been reported in the literature [19]. These cases were mostly middle aged and located in pelvic/retroperitoneal and deep soft tissue. Cases were often symptomatic and showed early metastases, especially to the lungs. Histopathologically, they were also more exaggerated than those of malignant SFTs and included severe pleomorphic epithelioid/round cell morphology, very high mitotic index (50/10 HPF), larger necrosis areas and heterologous components in dedifferentiated areas [18]. With the recognition of malignant and dedifferentiated SFT cases, the differential diagnosis spectrum has widened and the classical immunophenotype has begun to move away from being sufficient for definitive diagnosis. It may be difficult to differentiate malignant SFTs from synovial sarcoma (SS), dermatofibrosarcoma protuberans showing fibrosarcomatous features (F-DFSP) and dedifferentiated liposarcoma (DLPS). The classical immunofentotype of SFT is no longer considered very specific for SFT. Fortunately, a newly defined genetic aberration in SFTs contributed greatly to diagnosis [19,20]. It has been suggested that the increase in activation of STAT6 gene (a transcriptional factor resulting from the fusion of NAB2 and STAT6 genes mapped in chromosome 12) seen in 90-100% of SFTs, plays a primary role in the pathogenesis of SFT [18]. The STAT6 antibody is used as the most suitable immunohistochemical marker for the diagnosis of SFT with its high sensitivity and specificity [21]. Nuclear STAT6 expression is detected in almost all conventional and malignant SFTs, while STAT6 expression in dedifferentiated SFTs is lost [22]. STAT6 expression in the discrimination of SFT from SS, F-DFSP and DLPS is very helpful. Histomorphologically, SS which is similar to malignant SFT, is positive for TLE1 and can rarely show CD34 positivity [18]. TLE1 positivity may also be seen in SFTs. Therefore, it is important that SS is STAT6 negative. Because F-DFSP is typically a CD34 positive malignancy, it may be difficult to differentiate, especially with malignant SFT. F-DFSP is generally deep tissue localized and can be distinguished by minimal STAT6 expression. The situation for DLPS is a little more difficult because DLPS is positive for STAT6 in a rate of 11%. The determination of well differentiated LPS focus by expressions of MDM2 and CDK4 in DLPS will allowed discriminating from SFT [18]. Another diagnostic problem may be with dedifferentiated GISTs in which the losses of c-kit and DOG-1 expression can be observed. In such a case, mutation analysis in the genes of KIT and PDFGRA and the history of tyrosine kinase inhibitor treatment may be helpful in the differential diagnosis [18]. The tumor of our case had a diameter of 12 cm and it was above the threshold of 10 cm, which Shoubbege described as a risk factor. In our case, a retroperitoneal tumor with a pattern of patternless pattern beside hemangiopericytomatous pattern was observed. We reported our case as SFT with malignant histopathological features because of the presence of a high mitotic index (10/10 HPF), infiltrative border feature, large necrosis foci and vascular invasion. Although we examined the tumor of our case in many samples, we could not find any anaplastic/dedifferentiated focus. Our case demonstrated the characteristic immunophenotype of SFT. In our case, we found diffuse nuclear expression with PAX-8 (a member of the paired-box gene family expressed in renal, thyroid and Müllerian epithelial tumors) as an additional finding. Ulman, et al. reported that SFTs often showed PAX-8 expression, and PAX-8 expression was observed more frequently at extrathoracal locations [23]. It has been suggested that this might cause some problems in the differential diagnosis of retroperitoneal malignant tumors with sarcomatoid differentiation and might lead to serious diagnostic complications in especially sarcomatous renal cell carcinomas [23,24]. Despite diffuse PAX-8 expression in our case, the negative results for cytokeratin, CD10 and RCC marker were excluded the possible diagnosis of sarcomatoid renal cell carcinoma. In addition, the tumor of our case had no relation with the kidneys, radiologically. We have also ruled out other entities such as GIST, mesothelioma, angiosarcoma, liposarcoma, smooth muscle tumors, peripheral nerve sheath tumors, sarcomatous carcinoma and prostatic carcinoma, based on our histomorphological and immunohistochemical findings. Because of its variable and unpredictable clinical behavior, primary treatment in SFTs is en bloc surgical resection with negative margins. There is little evidence that adjuvant chemotherapy and radiation therapy following surgery is beneficial. Unresectable/metastatic SFTs do not respond to conventional therapies. Although treatments with anti-angiogenic drugs have shown promising results, the data is very limited. Dedifferentiated SFTs may be more sensitive than conventional SFTs to some chemotherapy regimens such as anthracycline based chemotherapy [18]. Long-term follow-up is recommended in all patients. The most important prognostic factor is the adequacy of surgical resection.
1. Malignant histopathological features and/or dedifferentiated areas in tumors with hemangioperitomatous pattern should not be omitted by careful histopathological examination.
2. An updated immunohistochemical panel including more specific antibodies for these cases should be applied.
3. In order to increase our knowledge about the clinicopathological features and biological nature of these tumors, there is a need more cases of extrathoracal/extrapleural SFT.
None.
The authors declare that they have no conflict of interest.