To demonstrate what are the main neurological dysfunctions within the hyperargininemia and other aspects of the disease in order to provide knowledgement and an update on the issue.
We conducted a literature search on reliable databases (PubMed/MEDLINE, Scielo/LILACS and UptoDate) from 1960 to 2018. The selection considered the most relevant articles, including 49 papers and 1 book for this narrative literature review.
Each of the selected materials was studied aiming to the formation of a cohesive and clear article. The main topics were sequenced in: clinical manifestations, diagnosis, genetics, and treatment.
Hyperargininemia is a rare and underdiagnosed disease, but it is benign due to unusual severe hyperammonemia. The main clinical signs are neurological, such as spasticity, ataxia, hyperreflexia, incoordination, paresis, bilateral Babinski sign, tremor and seizures. The initial suspicion occurs with retraction of the Achilles tendon and spasticity. The therapy focus into reducing plasma levels of arginine and maintain a normal ammonia plasmatic concentration.
Urea cycle, Arginase deficiency, Hyperargininemia, Guanidino compounds, Oxidative stress and its combinations
Arginine is an essential amino acid. This compound plays an important role in various body functions including cell division, wound healing, removal of ammonia, immune function, and release of hormones. The arginase 1 is a hepatic enzyme that is the faulty component in urea cycle. The urea cycle is represented in Figure 1 with focusing on arginine. When in normal function, the enzyme catalyzes the reaction of conversion of the arginine in ornithine and urea. However, in hyperargininemia, this enzyme is faulty and the concentrations of arginine in the plasma increases. The gradual accumulation of ammonia in plasma (hyperammonemia) is even more severe to the central nervous system (CNS) [1-3].
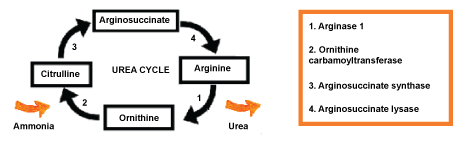 Figure 1: The enzyme arginase 1 catalyzes the arginine reaction in ornithine and urea. The ornithine carbamoyltransferase enzyme converts ornithine into citrulline. The enzyme arginosuccinate synthase transforms citrulline into arginosuccinate. This chemical component can be converted to arginine through the enzyme arginosuccinate lysase.
View Figure 1
Figure 1: The enzyme arginase 1 catalyzes the arginine reaction in ornithine and urea. The ornithine carbamoyltransferase enzyme converts ornithine into citrulline. The enzyme arginosuccinate synthase transforms citrulline into arginosuccinate. This chemical component can be converted to arginine through the enzyme arginosuccinate lysase.
View Figure 1
This disorder is uncommon when compared with other disorders of the urea cycle. The development of hyperammonemic encephalopathy is not observed in all cases. Other disturbs in the urea cycle can already be identified in the neonatal period or in the beginning of childhood just by the neurological clinical sign appearance. However, in the hyperargininemia, these signs generally do not appear prematurely [4,5].
Quantitative studies point to average prevalence rates of the disorders of the urea cycle more studied in the proportion of 1:8200 births in the United States. Therein, stands out that the hyperargininemia presents incidence rates of 1:2,000,000 live births [3,6-8].
The deficiency of the ornithine transcarbamylase enzyme presents higher rates of prevalence inside the disorders of the urea cycle. Yet the hyperargininemia and the deficiency of N-acetyl glutamic synthase are the less frequent disturbs of the group [3,9].
Although of the illness affect all ethnicities, a greater incidence of hyperargininemia is presented in the Canadian population. The first reported case of hyperargininemia occurred in 1969, with clinical findings of hyperargininemia and neurological dysfunctions such as lethargy, intellectual development disorder and seizures [10-12].
The fact that the hyperargininemia is less known than the other metabolic diseases of the urea cycle makes this disorder under diagnosed. Many individuals carrying the illness end up being diagnosed and treated late.
This illness is as important as the other metabolic illnesses in the urea cycle. The hyperargininemia requires medical attention to the main signs and symptoms that the bearers tend to present, in order to obtain an early diagnosis and offer the adequate treatment to these patients.
It was carried out a bibliographical research in trustworthy data base (PubMed/MEDLINE, Scielo/LILACS and up to Date) between the years of 1960 to 2018, using "Urea Cycle", "Arginase deficiency", "Hyperargininemia", Guanidino compounds", "Oxidative stress" descriptors and their combinations. The selection of articles considered the most relevant ones according to the coverage of the suggested theme; with that non-systematic, it was included 49 articles and 1 book as content to the literature review narrative.
From the selection of the most relevant materials to the scientific research about the main disorders that occurred in the hyperargininemia, each of them was studied with an objective to constitute an organized article in order to provide an update to the professionals in the health area. So, the main topics were sequenced in clinical manifestations, diagnosis, genetics, and treatment.
The main clinical signs are neurological. The illness shows the first symptoms in the childhood period. It is not common the detection of the hyperammonemia and encephalophaties in the newborn. The symptoms usually appear in the first two or four years of life. The progressive spasticity, the regression in the development and the reduction of the mental ability are very common [2].
In rare cases, from 3-months-old to 4-years-old of life, it may have the psychomotor deterioration, which is a sign of hyperammonemia. The hyperammonemia generates the deterioration of clinical symptoms gradually more significant [13].
The epilepsy and the progressive spastic diplegia are not common in newborns and children under 4-years-old. The newborns may present irritability and decrease in the alert state with the introduction of the bovine milk [9,11].
Cases of sharper hyperammonemia have already been reported, including, the abrupt evolution and fatal outcome. In a newborn patient with high plasmatic concentrations of arginine and glutamine in the liquor, the hyperammonemia got intensified. This dysfunction generated cerebral edema and tachypnea with progression to death [15].
Other clinic presentation is the presence of seizures. Researchers reported cases of continuous partial epilepsies related to hyperargininemia [16]. However, the generalized tonic-clonic seizure shows as the most prevailing [14].
The severe spasticity is a common alteration in the hyperargininemia, possibly presenting relation with the skeletal abnormalities. There are relates of patients with this condition that received treatment with valproic acid. They developed encephalopathy hyperammonemia [16,17].
There is description of the decompensation of the clinical status in patients with hyperammonemia and hepatic lesions in the clinical presentation of a possible cirrhosis [18].
In the physical neurological exam, the most consistent findings relate to the involvement of the upper motor neuron. This presentation was present in up to 80% of the cases. The neurologists may note the loss of voluntary motor abilities and the hyperreflexia. The athetosis and ataxia are less frequent [14].
It was reported that the guadinic compounds caused considerable inhibitions of the sodium-potassium-ATPase pump in the cerebral cortex. This is associated to a harmful effect in the CNS [19]. The compounds as the arginine, N-acetylarginine, homoarginine and argininic acid lead to the generation of free radicals. These compounds consequently decrease the antioxidant defenses. They change the catalase enzyme, dismutase superoxide and peroxidase glutathione. These enzymes are the main enzymatic defenses in the brain, contributing to the neurological dysfunction [20,21].
A severe elevation of guanidine compounds presents potential neurotoxic. Tests in vivo and in vitro point to the possibility of inhibition of some inhibitory gamma-aminobutyric acid (GABA) neurotransmitter canals [21].
The induction of seizures may also occur by decreasing the fluidity of plasmatic membrane of the neurons in CNS [22]. Studies also suggest that arginine decreases the activity of the acetylcholinesterase and the kinase creatine [23,24]. This effect damages the respiratory chain and compromises the memory function [24].
The arginine reduces the hydrolysis of the ATP, of the ADP, the AMP and the activity of the butyrylcholinesterase in the blood [25-27]. These changes may be prevented by giving the L-NAME (L-NG-Nitroarginine methyl ester). This is an inhibitor of nitric oxide enzyme synthase, provoking the nitric oxide to be involved in these effects [28]. In more advanced cases of the illness, it may also occur cerebellar and brain atrophy. Moreover, it can occurs the attack in the posterior portions of the putamen nucleus [29]. These main clinical symptoms can be summarized in Figure 2.
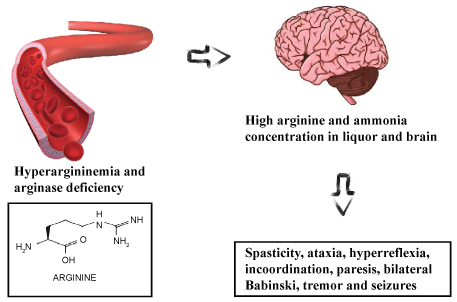 Figure 2: The Hyperargininemia generates high arginine and ammonia concentration in liquor and brain. This dysfunction generates the neurological symptoms.
View Figure 2
Figure 2: The Hyperargininemia generates high arginine and ammonia concentration in liquor and brain. This dysfunction generates the neurological symptoms.
View Figure 2
The diagnosis can be confirmed through laboratorial analyses with high concentrations of arginine in the plasma or in the liquor. This exam shows the defect of the arginase enzyme I. The plasmatic concentration of arginine may reach 1.5 Mm. There are data showing that in the cerebrospinal fluid, the values may be 10 times higher than the average value in plasma. The average plasmatic value varies from 21.8 to 87.8 µM and the average liquorice value is 6.8 µM [30-32].
The diagnosis may be confirmed in the pre-natal period. By the analyses of the amniotic fluid and chorionic villi, the genetic analyzes can be performed to find arginase 1 gene (ARG1) mutations [2].
One scientific article emphasizes the possibility and importance of establishing a national neonatal screening policy to ensure early detection of inherited metabolic disorders. This research suggest this goal in metabolic diseases which can be easily treated, such as hyperargininemia [33].
In this metabolic disturb, many components from the guanidine accumulate in the blood and in the liquor. The guanidine acetate is a major factor in the development of many physiopathology unbalance. This compound has an epileptic potential in CNS [34].
The nitric oxide and the homoarginine are also involved in the physiopathology of the illness. The nitric oxide, for example, is formed from the arginine under the nitric oxide synthase. It generates a free radical that may interact with the superoxide and bring to the formation of the peroxynitrite anion. This anion is highly cytotoxic [35].
With the development of the illness, the increase of level of guanidine compounds settle with the secondary biochemical path activation. The arginine, in high concentrations, is converted into α-acetic--guanidine acid valeric by transamination. This compound forms the arginine acid by hydrogenation. The arginine may also be converted into N-acetyl arginine by acetylation and acetic guanidine acid, β-guanidine butyric by transamidination. Yet the homoarginine, may be formed from the lysine by guanidination [3,35].
The magnetic resonance image (MRI) of the brain may show abnormalities. The main dysfunctions are the brain and cerebellar atrophies. Hyperargininemia can generates lesions in posterior portions of the putamen nucleus. These lesions occur in patients with advanced stages of neurological lesions in hyperargininemia. It hardly ever occurs in patients with lighter degrees of CNS damage in this disease. The high relation choline/creatine may indicate a deposition of arginine in the nervous tissue [29,36].
The histological evaluation of extracted biopsies from the liver of the hyperargininemia patients tend to show a low degree of fibrosis. However, anatomopathological studies reveal that the patients who develop hyperammonemia tend to show a higher degree of hepatic fibrosis associated to the atrophy of the CNS [14].
The hyperargininemia is characterized by an autosomal recessive genetic origin linked to the chromosome 6 (in the long arm) and to the ARG1 gene. Without a screening test, right after the birth or as soon as possible, the diagnosis becomes more difficult. The illness presents an almost asymptomatic characteristic in the early years [37,38].
The genetic analyses by amplification of the chain reaction of the polymerases already makes it possible to find mutations in ARG1. A study identified five mutations in the ARG1. This laboratorial technique detailed the location of the genetic changes in the chromosome 6 on hyperargininemia [38]. Researchers identified mutations in the gene ARG1 in Brazil. The most frequent mutation in the Brazilian patients analyzed was the p. T1341 [39].
Besides the main gene studied, there is the ARG2 gene. When it is expressed, it produces the formation of arginase enzyme II. This enzyme is found in the kidneys and prostate. However, even with the similar bimolecular characteristic, the function of this enzyme is still not fully understood. The relation of its deficiency and the appearance of the hepatic steatosis is submitted to more evidences. It is now not known if the mutations unitarily in the ARG2 gene may lead to hyperargininemia [40,41].
Experimental studies suggest that the low levels of arginase 1 (as low as 10%) would already be enough to avoid the plasmatic unbalance of hyperargininemia. This could provide a survival of the patients [42].
The gene therapy appears to be promising and useful to avoid the development of the illness, as its neuropathological effects. Arginase 1 gene therapy using adeno-associated virus rescued nearly all these abnormalities when administered to neonatal homozygous knock-out animals. Therefore, gene therapeutic strategies can reverse physiological and anatomical markers of arginase 1 deficiency and therefore may be of therapeutic benefit for the neurological disabilities in this syndrome. With neonatal administration of adeno-associated virus expressing arginase, there is near-total recovery of the abnormalities in neurons and cortical circuits [43].
In 2012, it was developed a learning algorithm, which selects sequences of an informative gene database. This algorithm shows the synthesis of the genes selected. It was created a set of informative of seven chimeras enzymatically active, with portions containing mutation close to the original arginase. This study provided greater understanding about the stability in the long term of the arginase 1. The arginase 1 deactivation in physiological conditions was better understood and its possible therapeutic use in the hyperargininemia patients [44].
The treatment consists in a controlled diet. The diet limits the excess of arginine. The frequent medical support is also important. The administration of pharmacological treatment (like benzoate and phenylbutyrate) brings benefits. Some patients present reduction of argininemia. Not all patients respond well to the treatment, but a relevant portion present efficient decrease in the neurological damages and improvement in the clinical state [2].
Patients treated and supervised since birth with controlled and limited protein diet and with supplementation of essential amino acids tend to present practically asymptomatic. The clinical manifestation is more evident in comparison to the patients who had the diagnostic for more than 30 years [45].
There are few reports of patients who needed the withdrawal of the nitrogen. This treatment is administrated by intravenous route after acute neurological symptoms in hyperargininemia patients. This portion represents a minority among the all patients [46].
An alternative treatment to the disturb is the use of drugs which increase the renal excretion of nitrogen by hippuric acid or phenylacetylglutamine. These options can be cited by sodium benzoate (250-375 mg/Kg/day), L-carnitine (100 mg/Kg/day) and even glycerol phenylbutyrate. It has been demonstrated that the treated patients with this drugs since birth, showed asymptomatic for long periods [47].
In a pediatric study, it showed the effectiveness potential of sodium benzoate given by seven months to a diagnosed child with hyperargininemia. This patient presented spastic paraparesis, ataxia and electroencephalograph abnormalities. The child presented improvement and stabilization of the levels of arginine and ammonia. The patient had a light cognitive deterioration [48].
Patients with secondary seizure to hyperammonemia in the hyperargininemia were treated efficiently with anticonvulsants. These drugs are an efficient option in this disease [16].
The available treatment is a first option to the stabilization of the disease. The reduction of the neurological symptoms and the decrease of the levels of arginine and ammonia are essential [49,50].
The hyperargininemia is an innate dysfunction of the urea cycle caused by the deficiency in the activity of the arginase 1. This enzyme is responsible of the conversion of arginine in urea and ornithine. This illness is biochemically characterized by high levels of arginine and arginine tissue accumulation of guanidino compounds.
The hyperargininemic patients may present a progressive state of spastic tetraplegia, growth restriction, seizures, hyperactivity and hepatomegaly. The neurological lesions generally occur in a progressive way. The patient can be diagnosed with spasticity, ataxia, hyperflexia, motor incoordination, paresis, bilateral Babinski sign, shiver and seizures.
Even being rare, the hyperargininemia is treatable and the therapeutic consists in reducing the levels of plasmatic arginine. The advisable treatment consists in poor diet of natural proteins (restricted in arginine). This approach is supplemented with sodium benzoate and L-carnitine, once the protein restriction isolated is not enough to uniform the levels of arginine.
Our expectation for the future is a further update on the disease by health professionals. Knowing how to perform the diagnosis and recognize early onset in children can change the patient's life of hyperargininemia. New researches for the development of a more effective treatment may be carried out in the future to bring better quality of life for patients with diseases such as hyperargininemia.
There is no conflict of interest in the present work and the authors did not obtain any financial support to its accomplishment.