The mammalian brain depends on a continuous supply of oxygen and glucose to meet metabolic demand. Adaptive mechanisms allow brain cells to exist under conditions of moderately low oxygen when 'idling' or exposure to high altitude as well as elevated oxygen delivery when activated. Regulation involves numerous intrinsic and extrinsic adaptive mechanisms that serve to maintain metabolic homeostasis. Under conditions of chronic low oxygen (10%) endogenous changes in CNS angiodynamics help promote survival. We questioned whether endogenous adaptation to a stressor such as mild changes in respiratory oxygen altered chronic inflammation in an animal model of multiple sclerosis (MS) experimental autoimmune encephalomyelitis (EAE). We previously reported that when mice were exposed to chronic mild hypoxia at the time of immunization with myelin oligodendrocyte glycoprotein (MOG) they exhibited a delay in disease onset and a decrease in disease severity. Changes in clinical EAE were associated with decreased T cell infiltration into the spinal cord as well as altered regulatory T-cell subsets. In this study, we examine the effect of in vivo exposure to chronic mild hypoxia after the development of disease symptoms. Mice were exposed to 10% normobaric oxygen after the clinical symptoms reach to score 2. Exposure to mild hypoxia ameliorated chronic inflammatory activity and was associated with reduction in clinical score. Further we found evidence that exposure to hypoxia increased the ratio of Foxp3+ Tregs and IL-10+ and Blimp1+ T cells following exposure to low oxygen. In addition, chronic low oxygen decreased evidence of endothelial activation.
EAE, Chronic hypoxia, FoxP3+ Tregs: IL-10+ T cells, Blimp1+ T cells, Endothelial activation, VCAM, Acclimatization and progressive EAE, Chronic inflammation
Homeostasis is a fundamental property of biological systems preserving stability by maintenance of key variables within an acceptable range [1]. In the brain, maintenance of homeostasis is the result of a coordinated effort between the cellular constituents of the neurovascular unit (NVU) [pericytes, endothelial cells, astrocytes and neurons] [2-4]. These cells make fine-tuned regulatory adjustments that promote tissue survival, maintain the balance between oxygen and glucose availability and neuronal metabolic demand. Adaptive mechanisms that maintain homeostatic balance reflect a continuous matching of tissue oxygen with capillary density and promote survival [5,6]. We have studied an in vivo model system that mimics the adaptive responses induced in the brain in response to a chronic mild hypoxic stimulus (10% oxygen) [5-8]. In vivo acclimatization to chronic mild hypoxia in animals is comparable to that achieved by humans when they encounter moderate high altitude [2,7,8]. Animals mount both systemic as well as intrinsic tissue and cellular adaptive processes that promote survival. Adaptation involves induction of angiogenesis and stabilization of normal vascular function.
In Multiple sclerosis (MS), it has been suggested that loss of bioenergetic homeostasis [9,10] may play a role in the initiation of axonal degeneration [10,11], and in turn may induce sensitization of the immune system [12]. This would suggest that therapeutic intervention aimed at restoration of vascular function and metabolic homeostasis may have a profound effect on tissue plasticity and disease outcome in both MS and EAE. We have questioned whether mechanisms aimed at maintenance of homeostasis alter the signs and symptoms of myelin oligodendrocyte glycoprotein (MOG) induced EAE. We reported that acclimatization to chronic mild hypoxia ameliorates the signs and symptoms of encephalomyelitis [13]. Exposure of mice to 10% oxygen significantly delayed the onset of clinical disease as well as reduced disease severity [14]. Changes in clinical disease was associated with decreased evidence of pathological disease as well as changes in T-cell effector and suppressor function. While the exact mechanisms are still unclear, they are likely to be highly complex and may involve changes in both vascular function as well as the immune system [13-15].
To get a better idea of the potential pathways involved, we questioned whether exposure to chronic mild hypoxia initiated following disease development similarly altered evidence of chronic inflammatory activity in MOG EAE. In this study, mice were immunized with MOG and exposed to chronic mild hypoxia following the development of clinical symptoms. Evidence indicated that exposure to low oxygen decreased chronic inflammatory activity through multiple pathways. Decreased chronic inflammatory activity was associated with changes in T-cell subsets, as well as restoration of vascular quiescence.
All animal experiments were approved by the Institutional Animal Care and Use Committee, Wayne State University (WSU), and conducted in accordance with the WSU guidelines (protocol # A-08-09-15). Female C57BL/6 mice (6-8wk) (Jackson laboratories, Bar Harbor, MN) were immunized with (MOG) peptide 35-55, (final concentration = 200 µg in 100 µl) emulsified with complete Freund's adjuvant (CFA) containing dead 5 mg/ml Mycobacterium tuberculosis (Difco Laboratories, Detroit, MI) sc. in two injection sites at hind flanks of each mouse. All mice were intraperitoneally (ip) injected with 150 ml of pertussis toxin (300 ng) (List Biological Laboratories, Campbell, CA) immediately after immunization and 2 days later. Mice were evaluated on a daily basis for overt signs of illness and clinical signs of disease using a 5- point scoring system: 0 - no symptoms; 1 - Limp tail; 2 - Limp tail and hind limb weakness; 3 - Partial hind limb paralysis; 4 - Full hind limb paralysis; 5 - Moribund state, or death by EAE.
MOG-immunized and sham-immunized mice were housed at 10% oxygen in normobaric hypoxia chambers (BioSpherix, NY) calibrated to administer 10% oxygen for varying periods of time [13]. Oxygen levels are maintained by a controlling system that regulates administration of nitrogen. Normoxic animals were housed in cages on the bench top next to the chambers. Both sets of animals were exposed to low oxygen for 21 days. Induction of adaptative mechanism in response to hypoxia was confirmed by weighing animals and measuring the hematocrit (Htc) values. Blood was drawn into heparinized capillary tubes and centrifuged and measured by using standard Htc charts. Animals were scored daily for evidence of clinical disease using a five-point scoring scale as detailed above. Animals were weighed, and blood samples taken for hematocrit. Vascular density in the spinal cord was determined in immunized mice exposed to normoxic and hypoxic conditions. Changes in vascular density in EAE and in response to low oxygen have been reported in our previous publications [13,14].
At various time points animals were euthanized and following tissue perfusion with PBS, spinal cords were flash frozen and 4 micrometer sections were made using a cryostat (Thermo Scientific Microm HM505N). Sections following air-drying and fixation were blocked with appropriate 5% normal serum in PBS for 30 min. Then the sections were stained with goat anti-mouse VCAM antibody (Santa Cruz, CA). Anti-goat IgG-594 (Invitrogen, CA) antibody was used as a secondary to visualize under red filter. In a different set of experiments, isolated microvessels were stained for expression of endothelial activation markers. Primary antibody directed against E-Selectin, ICAM, or VCAM (all from Santa Cruz, CA) was incubated with tissue then the appropriate fluorochrome conjugated secondary antibody was added for 1 hr. Microvessels were visualized in association with nuclear stain 4′,6- diamidino-2-phenylindole (DAPI), and pictures were taken by immunofluorescence microscope Leica DMLB. Mean fluorescence intensity (MFI) was calculated using Image J software. A minimum of 30 coverslips/group/time point were quantified.
Pathological evidence of disease that was shown by the level of demyelination was determined by staining the 40 µm sections of flash frozen spinal cords with oil red O (Sigma-Aldrich, CO). Sections were air-dried for at least 30 min, then fixed with methanol for 5 min, washed with distilled water twice, treated with 100% propylene glycol for 5 min, and then soaked in oil red O for 8 minutes at 60 ℃. Excess stain was removed by washing the slides with 85% propylene glycol for 5 min followed by a wash with distilled water. Sections were then lightly stained with Gill-3 hematoxylin (Lerner Laboratories, CN) for 3 min to visualize nuclei. Rinsed with tap water and then distilled water and mounted with aqueous based mounting media. Pictures were taken under bright field and 5x magnification by Leica microscope.
Anesthetized mice underwent thorough intracardiac perfusion with PBS. Brains and/or spinal cords were collected, minced into small fragments, and pressed through a 70-µM mesh sieve into Hank's balanced salt solution (HBSS) containing 10% fetal bovine serum (FBS) before digestion with collagenase (0.2 mg/ml; Worthington Biochemicals) and DNase (28 U/ml; Sigma-Aldrich) for 40 minutes at 37 ℃. Following enzyme neutralization, cells were layered onto a discontinuous Percoll gradient (1.03 to 1.088 g/ml) (GE Healthcare Life Sciences) and centrifuged at 2,400 rpm for 20 min at room-temperature in a swinging bucket rotor. After centrifugation, myelin debris was carefully aspirated, and the cell interface collected. Following extensive washes and incubation in Fc BlockTM (BD Biosciences, San Diego, CA) to minimize non-specific antibody binding to Fc receptors, cells were re-stimulated in vitro with PMA (50 ng/ml; Sigma Systems) and ionomycin (1 mg/ml; Sigma Systems) for 5 h at 37 ℃ in the presence of brefeldin A (1x; BioLegend) for intracellular staining, and analysis of cytokine production. After stimulation, cells were stained with fluorochrome conjugated primary antibodies for surface makers (CD45, CD3, CD4, and CD25) and then fixed and permeabilized using a fixation/permeabilization kit (BD Biosciences) for 20 min at 25 ℃. The cells were dual stained with conjugated antibodies against FoxP3, IL-17, IFNγ, IL-10, and Blimp1. Cells were analyzed using a BD FACSAria with compensation set based on the staining of each individual fluorochrome alone and correction for autofluorescence with unstained cells.
Mouse spinal cord capillaries were prepared as described previously [16,17]. Briefly, spinal cord tissue was removed within minutes of decapitation using sterile technique. Tissue was homogenized in 10 volumes of Dulbecco's modified Eagle's medium (DMEM) using a glass homogenizer and teflon plunger shaved to leave 0.25 µm between the plunger and the glass surface. After 20 up-and-down strokes at 420 r.p.m., the homogenate was centrifuged, and the pellet resuspended in 17% dextran in DMEM. The suspension was centrifuged at 5,000 g for 10 mins (Sorval DuPont, Wilmington, DE, USA), and the pellet resuspended and filtered through 118 µm nylon mesh. The filtrate was passed through an 40 µm nylon mesh. Microvessels (MV) were collected on the 40 µm mesh, washed vigorously from the mesh, and resuspended in DMEM. Microvessel preparations were 80% to 100% capillaries. MV were > 95% viable by trypan blue exclusion.
MOG immunized mice were evaluated daily as detailed in methods. Mice were placed in hypoxia chambers when they had reached a clinical score of 2. At this stage animals will ultimately switch to chronic disease phase (Figure 1). Animals were observed daily for 3 weeks. Results indicated that exposure to chronic mild hypoxia significantly reduced clinical scores during the chronic phase of the disease (Figure 2a). To visualize demyelination sections were stained using red oil O. Results indicated that evidence of demyelination was reduced in the spinal cords of hypoxic EAE compared to normoxic EAE mice after 40 days following immunization (Figure 2b). Reduced demyelination has associated with the lower clinical scores in the hypoxic EAE mice.
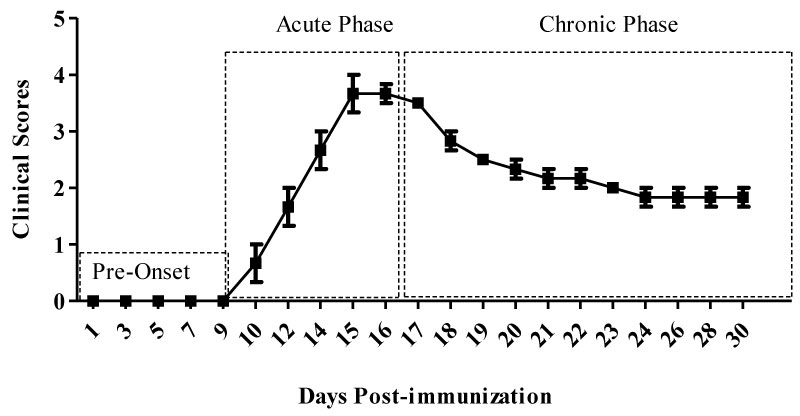 Figure 1: Clinical phases of MOG-induced EAE. Following MOG-CFA emulsion and PT injections animals are symptom free during the first 8-10 days, which we define as the pre-onset phase. Once the symptoms start they progress and reach to a peak between 14-16 days (acute phase), then the animals may recover to some degree and begin what we have termed (chronic phase). View Figure 1
Figure 1: Clinical phases of MOG-induced EAE. Following MOG-CFA emulsion and PT injections animals are symptom free during the first 8-10 days, which we define as the pre-onset phase. Once the symptoms start they progress and reach to a peak between 14-16 days (acute phase), then the animals may recover to some degree and begin what we have termed (chronic phase). View Figure 1
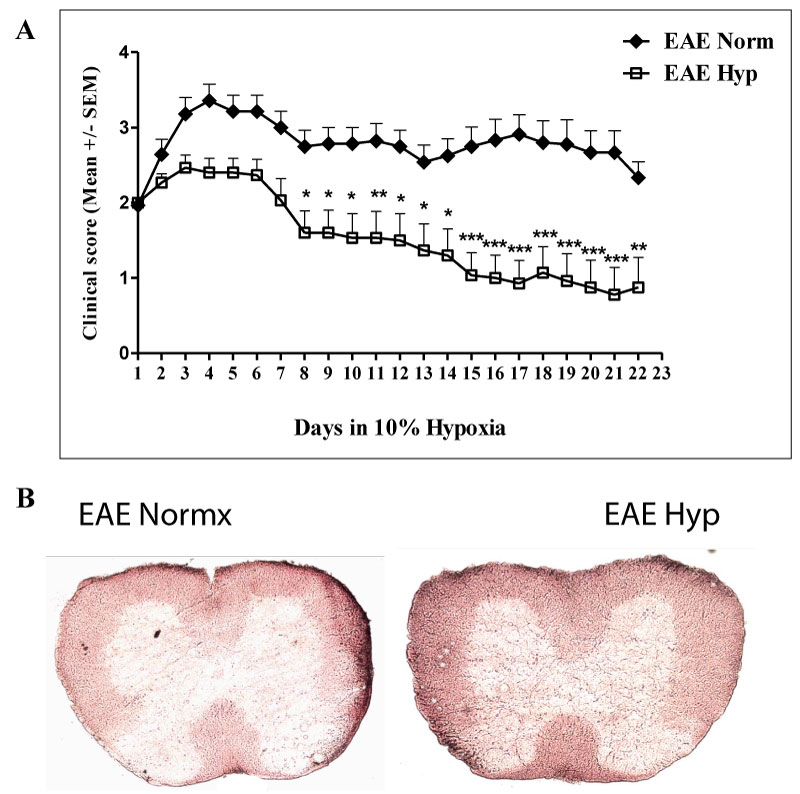 Figure 2: Exposure to chronic mild hypoxia decreased the inflammation and the severity of symptoms. Mice were immunized with MOG35-55 peptide + CFA. Once they reached to the clinical score of 2, they were placed in the normobaric hypoxia chambers at 10% oxygen for 3 weeks. The normoxic EAE group was left just outside the chambers for the same period of time. They were scored daily, and after 3 weeks of hypoxia all mice along with the normoxic group were sacrificed: a) The graph presents the Mean ± SEM of scores (n = 14-15 mice/ group) from 4 independent experiments. The two-way ANOVA analysis revealed that the difference was statistically significant (p < 0.001). Post-test Bonferroni revealed the significantly different time points; *p < 0.05, **p < 0.01, ***p < 0.001; b) Flash frozen spinal cords were prepared for red oil O staining as described in the materials and methods section to visualize demyelination. View Figure 2
Figure 2: Exposure to chronic mild hypoxia decreased the inflammation and the severity of symptoms. Mice were immunized with MOG35-55 peptide + CFA. Once they reached to the clinical score of 2, they were placed in the normobaric hypoxia chambers at 10% oxygen for 3 weeks. The normoxic EAE group was left just outside the chambers for the same period of time. They were scored daily, and after 3 weeks of hypoxia all mice along with the normoxic group were sacrificed: a) The graph presents the Mean ± SEM of scores (n = 14-15 mice/ group) from 4 independent experiments. The two-way ANOVA analysis revealed that the difference was statistically significant (p < 0.001). Post-test Bonferroni revealed the significantly different time points; *p < 0.05, **p < 0.01, ***p < 0.001; b) Flash frozen spinal cords were prepared for red oil O staining as described in the materials and methods section to visualize demyelination. View Figure 2
Our previous study has suggested that adaptation to hypoxia initiated before the clinical symptoms has decreased the number of infiltrating cells into the spinal cords [14]. Do determine if that is the case when hypoxia exposure starts after the disease onset, we first determine the total number of infiltrating T cells. Our results have shown that total numbers of infiltrating CD45+ and CD3+CD4+ immune cells were not significantly different between the EAE induced mice that were exposed to hypoxia after they scored 2 compared to normoxic EAE (Figure 3). Similarly, CD8+ T cells which accounted less than 1% of total T cells were not different among the hypoxia and normoxia groups either.
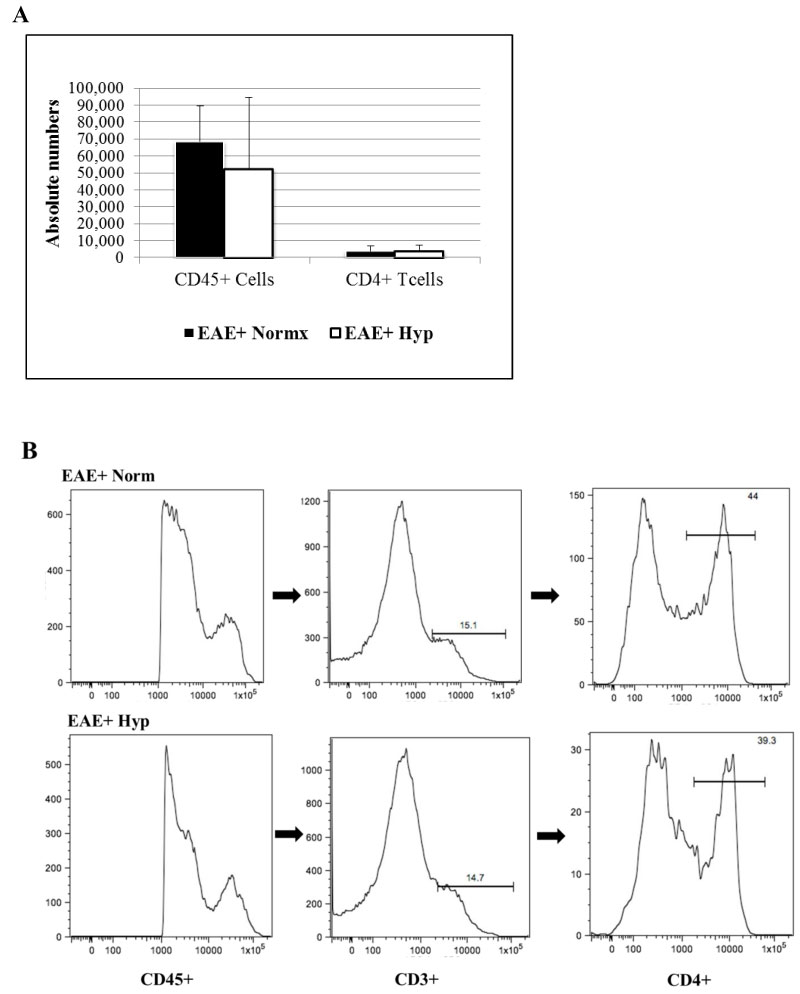 Figure 3: Leukocytes were recovered from individual spinal cords of MOG immunized mice (5-6 animals/group) after a 3-week exposure to chronic mild hypoxia. Mice were placed in the chambers when they reached a clinical score of 2. Leukocytes were immuno-stained and analyzed by flow cytometry: a) Bar graphs represent the absolute numbers of isolated leukocyte subsets (Mean ± SD); b) Histograms show the gating, and the flow-cyometric analysis of leukocytes isolated from the spinal cord. All (i.e. both high and low) CD45+ populations are further gated for CD3+, and subsequently for CD4+ populations. No significant difference in absolute numbers of CD4+ T cells was seen. View Figure 3
Figure 3: Leukocytes were recovered from individual spinal cords of MOG immunized mice (5-6 animals/group) after a 3-week exposure to chronic mild hypoxia. Mice were placed in the chambers when they reached a clinical score of 2. Leukocytes were immuno-stained and analyzed by flow cytometry: a) Bar graphs represent the absolute numbers of isolated leukocyte subsets (Mean ± SD); b) Histograms show the gating, and the flow-cyometric analysis of leukocytes isolated from the spinal cord. All (i.e. both high and low) CD45+ populations are further gated for CD3+, and subsequently for CD4+ populations. No significant difference in absolute numbers of CD4+ T cells was seen. View Figure 3
We then questioned whether there were alterations in CD4+ T-cell subsets among groups. It is well established that encephalitogenic T cells, especially the IL-17+ T cells play a major role in the pathogenesis of EAE [18]. While the presence of CD4+ cells in the spinal cord is highly correlated to disease severity [14,19], the presence of regulatory T cells [CD25+FoxP3+ regulatory T cells (Tregs)] can dampen neuro-inflammation. We examined the effect of chronic mild hypoxia on regulatory T cell subsets (FoxP3 expressing CD25+ T regs, Blimp1+, and IL-10+ CD4 T cells) along with encephalithogenic T cells (IFNγ+ cells, and IL-17+ cells). Our results showed that the percentages of FoxP3 expressing CD25+ T cells, IL-10+ T cells, and Blimp-1+ cells were increased significantly in the spinal cords of EAE mice exposed to hypoxia after disease onset (Figure 4). However, the IFNγ+ or IL-17+ T cells were not significantly different among the groups (Figure 4). These data suggest that induction of regulatory T cells in mild hypoxic conditions may overcome the detrimental effects of encephalithogenic T cells, and demyelination and the clinical scores recover to some degree despite the presence of encephalithogenic T cells.
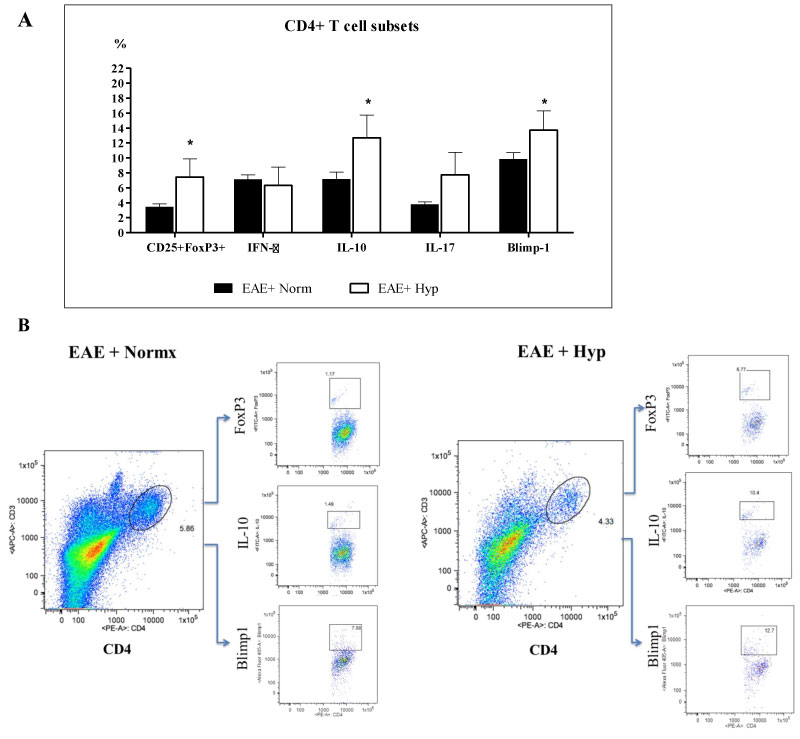 Figure 4: Leukocyte regulatory subset analysis. Exposure to chronic mild hypoxia after developing a clinical score of “2” caused significant increase in FoxP3+, IL-10+ and Blimp1+ T cells in the hypoxic EAE mice compared to those kept at normoxic conditions. Immune cells were recovered from the spinal cords of mice (5-6 animals/group) at day 35 following MOG immunization as described in the materials and methods section and analyzed by FACS. Results are presented as the percent of positive cells for each population, and the graphs: a) Represent the Mean ± SEM of three independent experiments. Student's T test revealed the significant differences between the normoxic and hypoxic EAE groups, *p < 0.05; b) The dot plot figures show the gating, and the representing population of FoxP3+, IL-10+ and Blimp1+ T cells in each group. View Figure 4
Figure 4: Leukocyte regulatory subset analysis. Exposure to chronic mild hypoxia after developing a clinical score of “2” caused significant increase in FoxP3+, IL-10+ and Blimp1+ T cells in the hypoxic EAE mice compared to those kept at normoxic conditions. Immune cells were recovered from the spinal cords of mice (5-6 animals/group) at day 35 following MOG immunization as described in the materials and methods section and analyzed by FACS. Results are presented as the percent of positive cells for each population, and the graphs: a) Represent the Mean ± SEM of three independent experiments. Student's T test revealed the significant differences between the normoxic and hypoxic EAE groups, *p < 0.05; b) The dot plot figures show the gating, and the representing population of FoxP3+, IL-10+ and Blimp1+ T cells in each group. View Figure 4
To further investigate the potential mechanisms that may be involved in mild hypoxia-induced alteration of immune cell infiltration in EAE, we questioned whether BBB permissiveness as defined by endothelial activation was altered. We first examined section of lumbar spinal cord for expression of the endothelial activation marker VCAM. Results in Figure 5a indicate that expression of VCAM was decreased in the sections derived from animals exposed to low oxygen. However, visualization and quantification is difficult in stained sections. We therefore isolated microvessels from spinal cord of treated and non-treated animals in order to more clearly visualize endothelial cell (EC) expression of activation antigens. Microvessels were isolated from the lumbar spinal cords of normoxic and hypoxic EAE mice at various time points and evaluated for the expression of endothelial activation markers, E-Selectin, ICAM, and VCAM (Figure 5b, Figure 5c, Figure 5d and Figure 5e). As expected, and previously published by our group endothelial activation is associated with development of EAE [14]. EC activation marker expression in the immunized EAE mice exposed to hypoxia was decreased after 21 days (Figure 5b). The mean Fluorescence intensity (MFI) of microvessels isolated from hypoxic and normoxic vessels on various time points in the hypoxic environment was quantified using NIH Image J software (Figure 5c, Figure 5d and Figure 5e). Exposure to chronic mild hypoxia after disease onset diminished activation marker expression steadily in a time dependent manner (Figure 5c, Figure 5d and Figure 5e). Vascular density was increased over the 3-week period. Taken together, our results suggest that endogenous adaptation to chronic mild hypoxia decreases endothelial activation and restores vascular quiescence.
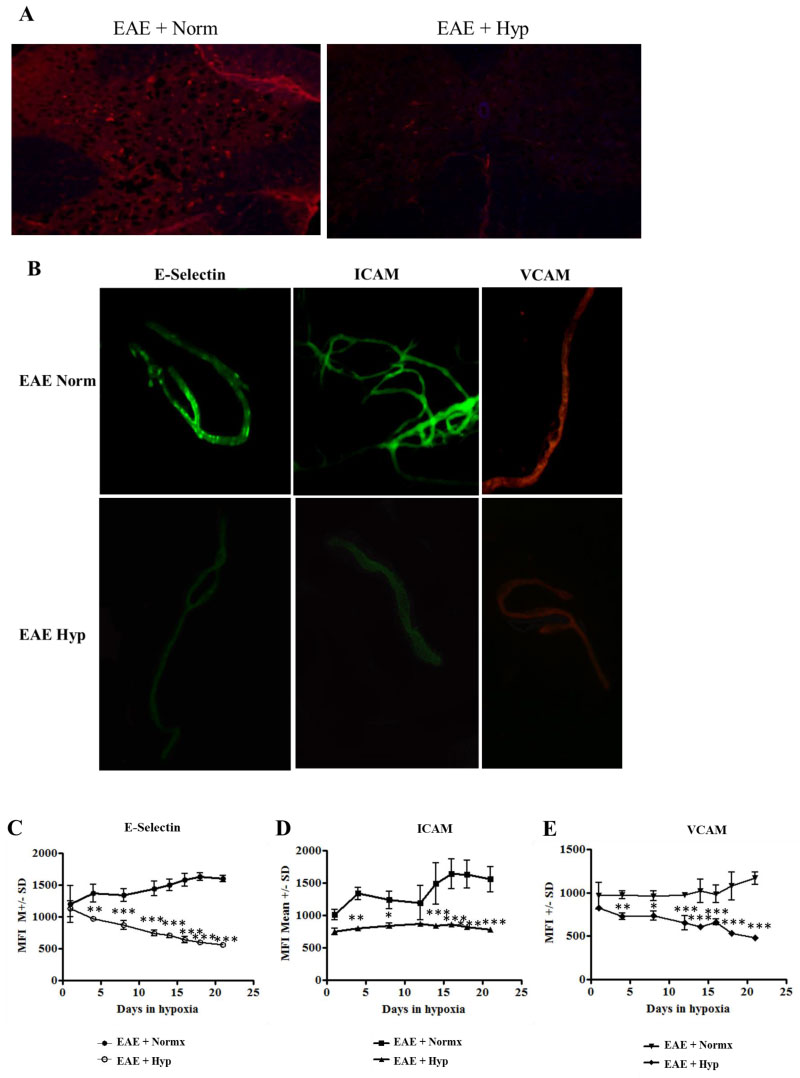 Figure 5: Evidence of vascular stabilization. EAE is associated with development of a permissive blood brain barrier. Permissiveness is concomitant with upregulation of adhesion molecules in the luminal surface of the endothelium [50]: a) Exposure to chronic mild hypoxia attenuated evidence of endothelial activation. Spinal cord sections were stained with antibody directed against VCAM as an initial indicator of activation. In order to quantify changes in endothelial activation we examined expression of endothelial activation markers in isolated microvessels. Spinal cords were flash frozen from MOG immunized mice at various time points following exposure to mild hypoxia or to normal oxygen levels; b) Microvessels were isolated from the spinal cord and stained for expression of endothelial activation markers E-Selectin, ICAM, and VCAM; c, d, e) Stained vessels were analyzed and MFI determined using NIH Image J software. Graphs represent the Mean ± SD of at least 30 coverslips of microvessels (n = 3 mice/group/time point). Asterisks denote the statistically significant differences; *p < 0.05, **p < 0.01, ***p < 0.001. View Figure 5
Figure 5: Evidence of vascular stabilization. EAE is associated with development of a permissive blood brain barrier. Permissiveness is concomitant with upregulation of adhesion molecules in the luminal surface of the endothelium [50]: a) Exposure to chronic mild hypoxia attenuated evidence of endothelial activation. Spinal cord sections were stained with antibody directed against VCAM as an initial indicator of activation. In order to quantify changes in endothelial activation we examined expression of endothelial activation markers in isolated microvessels. Spinal cords were flash frozen from MOG immunized mice at various time points following exposure to mild hypoxia or to normal oxygen levels; b) Microvessels were isolated from the spinal cord and stained for expression of endothelial activation markers E-Selectin, ICAM, and VCAM; c, d, e) Stained vessels were analyzed and MFI determined using NIH Image J software. Graphs represent the Mean ± SD of at least 30 coverslips of microvessels (n = 3 mice/group/time point). Asterisks denote the statistically significant differences; *p < 0.05, **p < 0.01, ***p < 0.001. View Figure 5
However, since the activation infiltration of immune cells start during the pre-onset phase of the disease, exposure to hypoxia after the onset of clinical symptoms can not affect the infiltrated cell number but the phenotype of cells is affected towards more anti-inflammatory cell type.
In the brain bioenergetic homeostasis is the result of a finely tuned process maintained by the cellular constituents of the NVU [2-4]. Maintenance of metabolic balance is essential for the survival of neurons and the other cells within the CNS. To achieve balance endogenous mechanisms have been developed to continuously match oxygen demand and supply by adjusting capillary density. As such, in response to stress stimuli such as chronic exposure to mild hypoxia the brain undergoes a number of extrinsic and intrinsic adaptations that in the CNS results in structural adaptations including vascular remodeling. Brain tissue parenchyma exists in a low oxygen environment. Quantitative measurements of brain tissue PO2 by polarographic microelectrodes have shown that the distribution of oxygen tensions in brain is asymmetrical and shifted towards lower concentrations [7]. More than two-thirds of the PO2 values are below 2kPa, and 90% are below the venous PO2 partial pressure of 5 kPa. Decreases in inspired oxygen produce decreases in tissue PO2 and decreased capillary blood hemoglobin saturation also seen in EAE animals (unpublished data). This decrease occurs despite an increased blood volume and faster capillary mean transit times as a result of vasodilation and increased cerebral blood flow [8]. The blood flow increase in response to mild hypoxia is at least partially mediated via oxygen-sensing neurons in the rostral ventrolateral medulla [9]. With initial exposure to hypoxia there is an increase in ventilation rate. This is driven by the carotid body chemosensory pathway through brain stem nuclei. The ventilation rate increases acutely then is maintained. Hyperventilation results in a decreased PCO2 (partial pressure of carbon dioxide) which allows the arterial oxygen tension to rise equivalently. The other adaptation seen at the tissue level and has a slower onset occurring only after days/weeks of exposure to 10% oxygen [10]. If mild hypoxic exposure is maintained, HIF-1α is induced, and angiogenesis is initiated, which results in an increased capillary branching and density. This produces decreased inter-capillary distances and thus decreased diffusion distances, restoring tissue oxygen tensions to baseline and tissue survival.
Endogenous adaptation to stress stimuli has been shown to promote tissue plasticity [6,20]. We have questioned whether endogenous adaptive mechanisms induced in response to mild stress alter inflammatory mechanisms in EAE [14]. Both sham immunized and MOG immunized animals responded to mild hypoxia by an increase in expression of HIF-1, and the HIF-1 dependent genes such as VEGF [14]. Both groups responded to the stress stimulus with an increase in vascular density [14]. However, the induction of the stress response to mild hypoxia also has a profound effect on the inflammatory response [14]. Exposure to mild hypoxia significantly delayed the onset of the signs and symptoms of clinical disease when animals were placed in the hypoxia chambers at the day of immunization. There was a clear reduction in cellular infiltrates seen by histology of lumbar spinal cord sections. Delayed induction of disease correlated with a delay in CD4+ leukocyte infiltration and Th17 specific cytokine responses [14]. We found that CD4+ cells were significantly decreased in the spinal cords of EAE mice exposed chronic mild hypoxia when compared to normoxic EAE mice. This was particularly apparent in MOG-specific Th17 responses. Therefore, exposure to low oxygen appeared to alter a key element in EAE pathogenesis. For many years, IL-17 secreting cells are considered a critical effector population in autoimmune demyelinating disease. IL-23 polarizes IL-17 secreting myelin-reactive CD4+ T cells. These cells are highly encephalitogenic upon transfer into naïve syngeneic recipients [18,21,22]. More recently, it has been shown that there was a resistance to autoimmune demyelination in mice reconstituted with CD4+CD25+FoxP3+ regulatory T cells [19,23,24].
In this study animals were not exposed to hypoxia until after clinical symptoms appeared. At this protocol, hypoxia exposure after the clinical symptoms attenuated the endothelial activation markers however it did not affect the total number of infiltrated encephalithogenic (IFNγ+ or IL-17+) T cell populations, probably due to the fact that infiltration of immune cells into the CNS has already started during the early phases of EAE. On the other hand, the regulatory T cells like FoxP3+ and IL-10+ T cells that have anti-inflammatory activity [25,26] were found increased in the hypoxia exposed EAE mice. Similarly, hypoxia exposure also increased the number of Blimp1+ T cells in the spinal cords of MOG-immunized mice. Previously, we have reported that chronic mild hypoxia itself was inducing the IL-10 cytokine levels in the CNS, and its levels were higher in the hypoxia exposed EAE mice than the normoxic EAE animals [14]. A supportive data related to the effect of hypoxia on IL-10 came from a study by Clambey and co-workers [27] showing that IL-10 can be induced in T cells under hypoxic conditions in in vitro. Production of IL-10 following immune cell recruitment into sites of inflammation limits macrophage and dendritic cell activation and maturation, resulting in suppression of sustained T cell function [26,28]. Beside IL-10, Blimp-1 is also involved in the regulation of T cell subsets. It is required for the differentiation of B cells into plasma cells [29], and T cell Blimp1 plays a critical role in protection against autoimmune encephalomyelitis through differential modulation of the effector functions of different T cell subsets such as cytotoxic T cells, Th17 cells, and T follicular helper cells [30-34]. Mice with a T-cell-specific deletion of Blimp1 [33] show a dramatic expansion of the effector T-cell compartment, increased capacity for homeostatic expansion and severe T-cell-mediated immune pathology. In addition, Blimp1 has also been implicated in IL-10 production from Tr1 cells [35,36] and the production of IL-10 by effector CD8+ and Foxp3+ regulatory T cells (Tregs) requires the expression of Blimp1 [35,37]. Taken together, these data suggest that infiltration of activated T cells into the CNS starts during the pre-onset phase of the disease, and exposure to hypoxia after the onset of clinical symptoms can not affect the number of infiltrating cells but skews the phenotype of them towards more anti-inflammatory cell type.
This anti-inflammatory milieu seen in hypoxia exposed EAE mice can be HIF-1α dependent since adaptation to mild hypoxia is under the regulation of HIF-1α. Increased expression of HIF-1α is first seen within 4-8 hours in vascular pericytes followed by astrocytes and oligodendrocyte progenitor cells [38]. HIF-1 has been shown to play a role in T cell differentiation [39], and especially in regulation of Tregs [39]. It was clearly shown that hypoxic conditions via HIF-1a induces transforming growth factor-beta (TGF-β) [40-42] which is the main transcription factor involved in the regulation of Th17 and Treg balance. In the absence of proinflammatory cytokines, TGF-β drives differentiation into Treg cells [43-45]. Therefore, CD4+ T cell subsets are not fixed; rather, plastic and they can change into other subsets when stimulated by different cytokines [46], and our results support this data. Comparable to previous studies [27,39,47] in our experiment's hypoxia has increased the ratio of FoxP3+ Tregs and IL-10+ T helper cells suggesting that adaptation to mild hypoxia favors the induction of regulatory T cells in EAE.
It is reported by several researchers that the development and severity of EAE is regulated by a balance between encephalitogenic and suppressor immune cells [19,23,24]. Since our results also imply the importance of this balance, we have suggested a working model related to the effects of chronic mild hypoxia in EAE (Figure 6). As we know from our and the other's previous studies the endothelium in patients with MS and in animals with EAE is activated [13,15]. As a consequence of this activation the expression of leukocyte adhesion molecules increase and inflammatory cell infiltration fosters [48-50]. When immunized animals were exposed to hypoxia following development of clinical symptoms, expression of endothelial activation markers was depressed. This effect could be mainly due to increase in TGF-β, HIF-1 and related genes which may also shift the balance of T cells and inflammatory milieu towards more regulatory T cells and anti-inflammatory mediators. As a conclusion, adaptation to mild hypoxia restores vascular quiescence at the BBB, inhibits endothelial activation markers, increases vascular density, and dampens inflammation. Consequently, metabolic homeostasis restores, and repair starts.
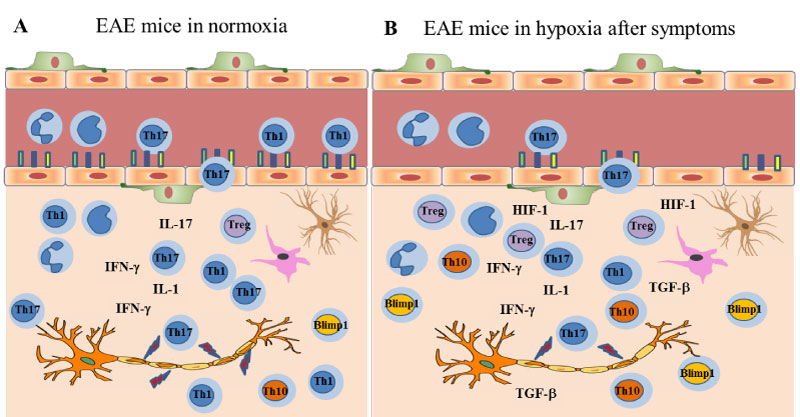 Figure 6: The inflammatory milieu in EAE: a) Normoxic conditions: During the pre-onset phase following MOG-immunization, T cells get activated, endothelial cells lining the BBB express activation markers which in turn help T cells, B cells and antigen-presenting cells (APCs), including macrophages, enter the central nervous system (CNS). Infiltrated and resident immune cells secrete inflammatory cytokines that damage myelin, and the clinical symptoms are observed; b) Exposure to chronic mild hypoxia: Once the symptoms start, exposure to chronic mild hypoxia still help with recovery by; 1- decreasing the endothelial activation markers, 2- inducing TGF-β, HIF-1 and related genes, 3- switching the balance of inflammatory T cells towards anti-inflammatory T cell subsets such as Tregs, Th10+, and TBlimp1+ cells, and thus limiting the progression of symptoms. View Figure 6
Figure 6: The inflammatory milieu in EAE: a) Normoxic conditions: During the pre-onset phase following MOG-immunization, T cells get activated, endothelial cells lining the BBB express activation markers which in turn help T cells, B cells and antigen-presenting cells (APCs), including macrophages, enter the central nervous system (CNS). Infiltrated and resident immune cells secrete inflammatory cytokines that damage myelin, and the clinical symptoms are observed; b) Exposure to chronic mild hypoxia: Once the symptoms start, exposure to chronic mild hypoxia still help with recovery by; 1- decreasing the endothelial activation markers, 2- inducing TGF-β, HIF-1 and related genes, 3- switching the balance of inflammatory T cells towards anti-inflammatory T cell subsets such as Tregs, Th10+, and TBlimp1+ cells, and thus limiting the progression of symptoms. View Figure 6
The authors wish to acknowledge funding from the National Multiple Sclerosis Society Collaborative Research Center Award.
N.E. and P.D.D. conceived and designed the experiments; N.E. performed the EAE experiments, flow cytometry staining and analysis; J.M. scored the animals and performed cryosectioning and immunohistochemistry; R.S. helped during tissue collection and staining; N.E. and P.D.D. wrote the paper.
The authors declare no conflict of interest.