Steroid immunosuppression has maintained a long-lasting relevance in renal transplantation. In addition to its role in preserving allograft survival, it is often the first line agent in the rescue treatment of acute rejection events. Its major drawbacks include metabolic adverse effects and long-term cardiovascular morbidities. Motivated by the need to avoid growth impairment, pediatric renal transplant community provided a template for steroid minimization strategy. Steroid sparing regimens are often successful in the context of induction therapy with lymphocyte depleting agents. Because most randomized controlled trials are conducted for duration of 1 to 5 years, it is unclear if steroid-based protocol confers a longer renal allograft life span. By avoiding early-onset rejection, steroid prevents exposure of immunogenic epitopes and therefore reduces the development of late-onset antibody-mediated allograft injury. In contrast to the calcineurin inhibitors, steroid promotes allograft tolerance by enhancing preservation of the regulatory T cells. It suppresses inflammatory response to ischemic reperfusion events as produced by transplant surgery. Its anti-inflammatory effects are most beneficial in patients with high immunologic risk profiles. Finally, future therapeutic approach may embrace careful selection of most suitable drug regimen for an individual by using bio-molecular resources for risk categorization.
In the last six decades, we’ve witnessed a revolutionary success in short- and medium-term survival rates of renal allograft. Greater understanding of transplant immunology has led to an innovative use of immunosuppressive agents (ISA). However, recent data suggest the inadequacy of current ISA (rather than drug-induced toxicity) is the major culprit for late-onset allograft loss [1]. This is reflected by the association of the latter event with de novo development of donor specific antibody. This observation calls for cautious interpretation of data from short-term clinical trials on steroid minimization strategies. Indeed decision on the modality of steroid use must be supported by adequate knowledge of its pharmacology in multiple clinical settings. Given the long history of its use, there are available excellent data sources that are suitable for such inferential deduction. Hence, this article will examine the biological basis of steroid effectiveness, the mechanisms for its common adverse effects and the scientific merit for its combination with other ISA. The goal is to provide readers with theoretical background to enable him / her appreciate the desirability of steroid use [or non-use] in renal transplantation. For an in-depth analysis of the role of other classes of ISA, readers are referred to previous excellent reviews by other authors [1,2].
historical context Steroid was a pioneering ISA for the first cohort of kidney transplant recipients (KTR) in the early 1960s [3]. Its combination with azathioprine (AZA) produced a very modest one-year graft survival rate, barely reaching 40 to 50% [3]. Subsequent introduction of calcineurin inhibitors (CNI), mycophenolate mofetil (MMF), rapamycin (RAP) and lymphocyte depleting agents led to one-year graft survival rate in excess of 90% [4,5]. This extra-ordinary feat was, in part, due to the vast improvement in both surgical and organ preservation technologies. However in the last two decades, the steady increase in life span of renal transplant has been driven principally by the attrition rate in the first year. Due to inadequate immunosuppression and/or drug-induced nephrotoxicity, long-term allograft survival has been less satisfactory [4]. Thus rate of deceased donor graft loss in the first year dropped from 20% in 1989 to 7% in 2008, but it remained steadily constant at 5-7% over the same period for those that survived beyond one year [6].
United States registry data showed that more than 25% of the patients who are discharged from hospital after the transplant surgery have had a successful discontinuation of steroids treatment [6]. Although there is no consensus on the nomenclature for minimization strategies, for simplicity we shall use the following definitions: i) Steroid free: A strict definition of a complete elimination would imply there is no steroid use for induction, maintenance or as a rescue therapy for acute rejection (AR). Such approach will result in over-reliance on lymphocyte depleting antibodies and thereby expose patients to avoidable side effects. For this reason, most patients with AR are often treated with pulse doses of steroids. ii) Steroid avoidance is defined as discontinuation of glucocorticoids (GC) within 7 days of transplant surgery. Duration of steroid treatment for less than 3 days produces more frequent delayed graft function [7]. iii) Steroid withdrawal: Early withdrawal refers to discontinuation of steroids use between 7 and 14 days. Late withdrawals are achieved at various time points after 14 days of treatment. Up to 3, 6 and 12 months of GC use have been reported. It is associated with greater risk of graft rejection [8].
To enhance immunosuppression, renal allograft recipients placed on steroid minimization protocols are often treated with induction agents [4-8]. Due to superior efficacy, anti-thymocyte globulin (ATG) is often preferred in high risk patients. Compared with alemtuzumab, it is associated with lower rate of AR, and a longer duration of allograft survival [9,10]. Non-depleting monoclonal antibody (basiliximab, an IL-2 receptor agonist) has a wider therapeutic window and is therefore the treatment of choice in low risk recipients [11,12]. Adjunct treatment with methyl prednisolone (5-10mg/kg) during surgery may produce crucial non-immunologic benefits [13]. It suppresses the release of pro-inflammatory cytokines (interleukin-6 (IL-6), IL-8, TNF-α) in response to ischemic reperfusion injury. Furthermore, partly due to the anti-inflammatory effect, peri-transplant steroid use has been associated with a shorter duration of post-surgery hospitalization [13].
Comparison of studies on steroid sparing regimen is often confounded by variation in induction strategy, steroid doses, drug combination, co-morbidity pattern, and the choice of clinical endpoints [8,14-16]. Although similar rate of long-term allograft survival is observed in meta-analysis data, patients treated with steroid sparing protocols often showed higher rate of ARE but a less frequent metabolic side-effect [8,14-16]. The first RCT was performed in Canada in the early 1980s. Patients were randomized to receive either CsA only or in a combination with prednisolone. There was a greater need for steroid rescue and a lower rate of graft survival in the (CsA arm) former [15]. Similarly, CsA and mycophenolate sodium were utilized as maintenance agents in FREEDOM trial [16]. Greater proportion (41%) of the group treated with steroid-free protocol required rescue therapy compared with those (29%) on the early-withdrawal regimen [16]. Consequently, standard maintenance immunosuppression comprises of two or more classes of drug. This approach permits the use of minimum effective dose of these agents while curtailing their serious side effects [4,17].
Calcineurin inhibitor is widely considered as the most effective ISA used for the prevention of allograft rejection. Attempts to avoid these agents often result in greater rejection episodes and lower graft survival [4]. Hence tacrolimus and MMF with [and without] steroids are the initial drug combination in most patients [17].
Calcineurin inhibitor allows the preservation of essential immune function by producing a selective de-activation of specific lymphocyte function. Its major drawback is a dose-dependent renal toxicity [4,18].Compared with CsA, tacrolimus (TAC) produces a more favorable cardiovascular profile [18,19]. In a meta-analysis, ARE was avoided in 12 recipients and graft losses were prevented in 2 out of every 100 patients after 1 year of using TAC instead of CsA [20]. Concurrent steroid use may potentiate the metabolic complications [21]. Although lower doses of CNI may minimize adverse effects in high risk patients, there may be need for larger doses of steroids to prevent rejection episodes [22]. On the other hand, steroid use may attenuate CNI toxicity by decreasing its tubular accumulation via the activation of P-glycoprotein, a multi-drug efflux system [23]. Similarly, proximal tubular expression of (steroid-inducible) cytochrome P450-3A5 enzyme correlated inversely with the histology markers of cyclosporine injury [24] (Table 1).
Table 1: Pharmacodynamic inter-relationship of steroids and contemporary maintenance immunosuppressive agents in renal transplantation. View Table 1
Because of its anti-proliferative effect, there was initial enthusiasm that sirolimus (SRL) use might prolong allograft survival. However, in SYMPHONY trial, treatment with SRL was associated with the highest rate of ARE [25]. In addition, it failed to improve graft survival when used as a late substitute for CNI [4,25,26]. Everolimus, a derivative of SRL, has a similar immunosuppressive efficacy in renal transplantation. Both are associated with side effects that include new onset proteinuria, pneumonitis, hepatotoxicity, thrombotic microangiopathy, hyperlipidemia and delayed wound healing [27,28]. The last two events may be exacerbated by concurrent use of high dose steroids [4,25-28].
A RCT of steroid minimization strategy in which there was exclusion of MMF showed a higher rate of ARE and a double-fold increase in the short-term graft losses [29]. Similarly, patients with single nucleotide polymorphism [SNP] for uridine diphosphate glucuronosyl-transferase (UGT2B7), the metabolizing enzyme for MMF, experienced higher rate of ARE [30]. Demonstrating its superior efficacy over AZA, patients placed on MMF had lower events of graft losses [29]. However, such advantage was abrogated in a protocol that combined CNI, steroids and AZA after induction therapy with ATG [31].In addition, AZA may be more cost effective than MMF. Theoretically, steroid combination with either MMF or AZA may attenuate their suppressive effects on the bone marrow. However, unlike steroid up-regulation of Foxp3 (+) regulatory T cells, MMF reduced the functional capacity for post-ischemic tissue repair [32]. Finally, a combined use of TAC and MMF may promote BK virus replication by aggravating functional impairment of cytotoxic CD8 (+) T-cell [33].
Belatacept, recently approved by United States Food and Drug Administration (FDA), is a CTLA4Ig fusion protein that blocks co-stimulatory activation of the CD28 receptors on T cells. It produces higher rate of early-onset ARE, greater preservation of 12-month graft function but a similar rate of 5-year survival [34]. Due to a favorable cardiovascular profile, it may be more suitable than CNI for a combined treatment with steroids [35]. Although phase III trial had initially suggested there is a higher incidence of post-transplant lymph proliferative disorder, subsequent meta-analysis of multiple studies disproved this conclusion [34-36].
In a given individual, categorization of the risk profiles may determine the appropriate selection of ISA. In majority of the studies, beneficial effects of steroids are most pronounced in high immunologic risk patients [37,38]. These patients are particularly susceptible to late-onset chronic graft rejection. On the other hand, minimization of steroids and CNI may be more suitable for those at higher risk for cardiovascular morbidity [36-38]. A prospective study of early steroid withdrawal showed greater events of acute rejection in African-American recipients of kidneys from poorly matched donors [39]. Similarly, there is a four-fold greater risk of developing recurrent glomerulonephritis in steroid free recipients. Finally, a second episode of rejection is more likely to occur in the absence of GC maintenance after a rescue steroid treatment of ARE [40].
Theoretically, treatment with steroid may also reduce the development of late-onset antibody-mediated graft injury. The latter may be elicited by an inadvertent (prior) exposure to immunogenic epitopes [1,41]. The functional cooperation between the T- and B-lymphocytes may in part explain this phenomenon. In this regard, alloantigen internalized by B-cells is degraded into smaller peptides which are then expressed on the cell surfaces by MHC class II molecules [42]. Consequently, the antigen-presenting B lymphocyte forms an immunological synapse with the cognate receptor of CD4+ T-helper cells. The T-lymphocyte in turn stimulates antibody production from the B cells while there is a parallel activation of CD8 cytotoxicity. By enhancing apoptosis of both T- and B- lymphocytes, steroids reduce the opportunity for this immunological interaction [42]. Furthermore, it promotes hypo responsive state by reducing the exposure of allo-antigen to B cells and by inducing a selective preservation of the regulatory T-cells [43].
Prednisone is metabolized to the active agent, prednisolone in the liver. Its metabolic clearance depends on both hepatic cytochrome P450 and intestinal P-glycoprotein systems. Drug interaction, old age and ethnicity may account for the random variation in the bio-availability of steroids in transplant recipients [44-51]. Furthermore, due to increased metabolic clearance from large fat mass and higher hepatic blood flow, unadjusted dosing of steroids may result in sub-therapeutic exposure in obese patients. For these reasons, the traditional use of one-size-fits-all steroid treatment may contribute to a sub-optimal efficacy.
In the future, in addition to demographic risk categorization, genetic, metabolic and immunologic profiles may identify individuals that are best suited for a class of ISA. Although validation studies are needed, higher expression of RC isoform of CD45 molecule (CD45RC) on the surfaces of CD8 T cells in pre-transplant patients was predictive of ARE [45]. Similarly, measurement of lymphocyte proliferation in response to (a given amount of) endogenous cortisol identified those who are likely to fail steroid withdrawal [46]. In addition, pre-transplant serum levels of soluble CD30 correlate with the greater incidence of early onset AR events [47]. However, due to the confounding effect of single nucleotide polymorphisms, there are inconsistent findings on the relationship between cytochrome P450 genotype and CNI pharmacokinetics [48]. Finally, a development of auto-antibody to cytochrome P450 may lower the metabolic clearance of susceptible drugs [49]. This scenario is more likely in transplant recipients with auto-immune disorders and should be suspected if there is a sudden [unexplained] decline in CNI trough levels. Further studies are warranted to determine if there is a modulatory role of steroids in this circumstance.
Due to utilization of a common metabolic pathway, steroid increases the clearance of both calcineurin inhibitors and sirolimus. To a lesser degree it enhances enzymatic clearance of MMF by activation of uridinediphosphate-glucuronosyltransferase and the multidrug resistance-associated protein 2 [50]. Hence adverse clinical outcome must be monitored anytime there is a change in the drug combination [50,51]. For example, rapid tapering of steroids may increase tacrolimus exposure in fast drug metabolizers (homozygous cytochrome P450 3A5 genotype) while there is slower rate of metabolism in those with obesity and/ or hepatitis C infection [51].
Cellular activity of steroid is mediated by both genomic and non-genomic mechanisms [52]. The latter is less robust but has a rapid onset of action. It is mediated by membrane bound receptors. Its effect is blocked by mifepristone [a receptor antagonist] but it is not affected by actinomycin D, a transcription inhibitor [53]. Activation of receptors on the surface of T-cell (TCR) is ineffective in the absence of multi-protein complex of membrane-bound GC receptor (GCR), heat-shock proteins (HSP), lymphocyte-specific protein tyrosine kinase (LCK) and FYN oncogene. Glucocorticoid binding of TCR causes dissociation of the cytosolic LCK-FYN-HSP-GCR complex and therefore prevents the activation of signal transduction (Figure 1).
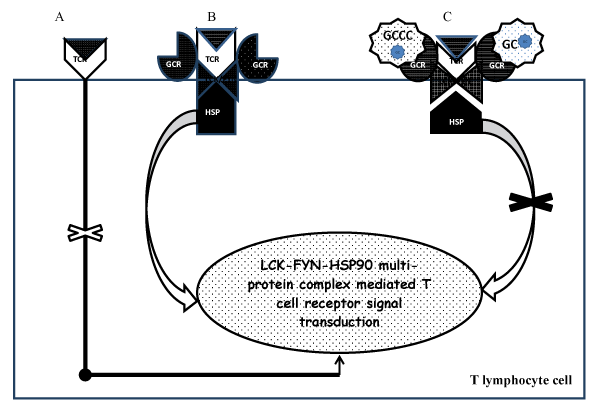 Figure 1: Molecular mechanism for the non-genomic inhibition of T cell receptor signaling system mediated by the GC activation of cell membrane-bound receptors.
Figure 1: Molecular mechanism for the non-genomic inhibition of T cell receptor signaling system mediated by the GC activation of cell membrane-bound receptors.
(A).Activation of T cell receptor (TCR) on cell membrane is ineffective in the absence of multi-protein complex that includes membrane bound GC receptor (GCR), heat-shock proteinschaperone system (HSP), lymphocyte-specific protein tyrosine kinase (LCK) and FYN oncogene (B). Successful activation of T cell receptor bound to multi-protein complex by a ligand (C). GC ligation of the membrane-bound receptor causes inactivation of the T cell signaling by producingthe dissociation of LCK-FYN-HSP-GCR complex.
View Figure 1
On the other hand, binding of glucocorticoid to the receptor stimulates phosphatidylinositol 3-kinase and protein kinase AKT. It activates endothelial nitric oxide synthase (eNOS) and inhibits the release of prostaglandin PGE2 [54,55]. Consequently, there is down-regulation of endothelial adhesion molecules, inhibition of neutrophil migration and prevention of phagocytosis. It minimizes enzymatic tissue injury by a reduction of the neutrophil secretion of elastase and collagenase; and suppresses the development of micro-vascular thrombosis by its inhibition of plasminogen activator [56]. Glucocorticoid depletes peripheral lymphocytes by promoting its sequestration within the reticulo-endothelial system but produces leucocytosis by systemic release of neutrophils from the bone marrow [57,58]. It inhibits lymphocyte activation and alters its action cytoskeleton by the dephosphorylation of ezrin-radixin-moesin protein [59,60]. This causes inhibition of the migration of effector T-cell and therefore reduces its interaction with prevailing allo-antigens [60]. Suppression of ezrin-moesin proteins may also decrease calcium influx which in turn reduces the intracellular signaling for IL-2 synthesis [61].
Genome-mediated cellular response has a slower onset but produces a more prolific biological activity. It involves translocation of cytosolic GC-bound-receptor into the nucleus where it binds steroid responsive elements (Figure 2). Inhibition of nuclear factor kappa B (NF-κB) repressed transcription of genes for the synthesis of pro-inflammatory mediators including IL-1, interferon-alfa [62], and inducible nitric oxide synthase (iNOS) [63]. Glucocorticoid inhibits both T- and B cell immune responses but produces a greater impact on T-cell function. Although universally expressed in most cells, glucocorticoid receptors are less pronounced in non-thymic cells. On the contrary, steroid treatment of the thymic lymphocytes produce accelerated apoptosis by induction of C3 isoform of GC receptor mRNA. Furthermore, double-positive thymocytes (CD4+CD8+) and natural killer T cells are more susceptible to glucocorticoid-induced cell death (GICD) [64].
 Figure 2: Simplified illustration of the cytosolic glucocorticoid binding system and translocation of the steroid-receptor complexes into the nucleus for the genemediated cellular function.
Figure 2: Simplified illustration of the cytosolic glucocorticoid binding system and translocation of the steroid-receptor complexes into the nucleus for the genemediated cellular function.
Extracellular glucocorticoid (GC) dissociates from steroid binding protein and crosses the cell membrane, facilitated by a relative small size and lipophilic property.
Inside the cell, maximum interaction with cytosolic glucocorticoid receptor (GR) occurs with the binding to a complex of chaperones that include immunophilin
binding protein (FK506-BP) and heat shock proteins (HSP). Activation of the GC-GR unit results in its dissociation from the multi-protein chaperones. Both
monomers and dimerized forms of GR-GC unitare translocated into the nucleus where they bind the responsive elements for transcription factors (TFRE) and the
glucocorticoid responsive elements (GRE) respectively. The transcription factors are activator protein-1 (AP-1), nuclear factor-κB (NF-κB) and signal transducer
and activator of transcription-5 (STAT5). The ultimate effects of this gene transcriptionsare inhibition of pro-inflammatory cytokines and downregulation of cellular
proliferation (cell cycle arrest/ apoptosis).
View Figure 2
Glucocorticoid treatment tilts the balance in favor of graft-tolerance by promoting the regulatory [Foxp3+ CD4+ CD25+] T-cells while inhibiting the cytotoxicity of T-effector cells [65,66]. Similarly, compared with the staining for IL-17 cytokines, individuals with renal histological specimen that expresses FOXP3 had lower frequency of steroid-resistant rejection events [66,67]. In addition, maintenance dose of steroids after a successful treatment of an acute rejection with pulse GC therapy results in a lower occurrence of a second episode [44]. On the contrary, by promoting a non-selective down-regulation of IL-2 receptor activity, CNI fails to preserve the tolerogenic effect of the regulatory T-cells [65].
Attempts to eliminate steroid and/ or CNI were motivated by the disproportionate impact of cardiovascular disease on the mortality rate of KTR with functioning graft [4,68]. Adverse effects of prolonged steroid use include hyperlipidemia, growth retardation, obesity, insulin resistance, hypertension, and bone diseases. By promoting apoptosis of plasmacytoid dendritic cells [PDC] and reducing the synthesis of interferon-alfa cytokine, steroid induction therapy increases the susceptibility of transplant recipients to opportunistic viral infection predominantly CMV and BK virus [69,70]. Steroid role in oxidative stress: While steroid (induction) modulates oxidative inflammation that results from IRI which is promoted by transplant surgery, its use as maintenance treatment in high doses may produce adverse carbonylation[catabolism] of the skeletal muscle protein [71,72]. This is partly due to activation of proteolytic ubiquitin-proteasome pathway which is induced by up regulating the expression of atrogin-1 and MuRF-1 genes [73,74]. Fortunately, such untoward effect is mitigated by endogenous elaboration of IGF-I which inhibits FOXO gene and decreases PI3-kinase signal transduction [74]. In addition, there is adaptive increase in the renal expression of tocopherol (α) transfer protein gene, which in turn promotes the recycling of anti-oxidative vitamin E [73,75].
Due to impaired glomerular filtration, pre-existing (donor) vasculopathy and vasocostrictive effect of CNI, hypertension is common in the first few days of kidney transplantation [76,77]. Loading dose of steroids as used for induction during transplant surgery may augment such hypertensive events. Excess intake of salt decreases endothelial synthesis of protective nitric oxide while raising the vasoconstrictive substance, asymmetric dimethyl arginine (ADMA) [76]. However, contrary to the theoretical expectation, steroid withdrawal protocol fails to attenuate the long-term prevalence of post-transplant hypertension. The principal mediator of steroid-induced hypertension is the glucocorticoid receptor activity [73]. This is demonstrated in an experimental mouse model of chronic steroid infusion that showed no significant change in the renal expression of mineral corticoid target genes including ENaC, Kras, and Nedd4 [73]. In addition, there was no evidence for tubular sodium reabsorption, potassium wasting or elevation in the plasma renin activity. Instead, GC effect increases the response to angiotensin II, which in turn enhances the synthesis of its (GC) receptors on vascular smooth muscle cell. It promotes hyper-filtration which causes an increase in the net sodium excretion [73]. Furthermore, there is (GC) reduction in the endothelial cell release of neuronal NO by the inactivation of protein kinase C signaling [78,79]. Finally, validating the effect of endothelial GC receptor, its knock-out mouse model failed to develop hypertension in response to dexamethasone infusion [80].
Due to the lower doses of steroid used in recent times, there is a falling incidence rate of new-onset diabetes mellitus after transplantation (NODAT) [81]. A study showed there is 5% risk of developing diabetes mellitus for every 0.01 mg/kg/day increase in prednisolone dose [82]. Similarly, there is 42% greater risk of developing NODAT in patients placed on steroid-based regimen over a period of three years [83]. Most likely there are additive effects from other agents. Hence calcineurin inhibitor, sirolimus and steroids are estimated to account for 74% incidence of NODAT. New onset diabetes mellitus has been associated with poorer graft survival and a three-fold higher risk of sustaining adverse CV events [84].
Predisposing factors of NODAT include genetic susceptibility, deceased organ donation, older age (>40 years), male gender, ethnic minority, exogenous obesity, hyperlipidemia, hypertension, hepatitis C infection, previous transplants, and ≥3 HLA class 1 mis-matches [85]. Similarly, greater number of metabolic syndrome components correlates with the probability of developing NODAT. Hence pre-transplant screening for the detection of modifiable metabolic events is an appropriate preventive strategy. Unfortunately, renal allograft survival is often limited by the occurrence of chronic immunologic injury that promotes a pro-inflammatory milieu which in turn increases susceptibility to metabolic disorders. This relationship is demonstrated in transplant patients with single nucleotide polymorphism for gene encoding interferon gamma cytokines and higher incidence of steroid-induced insulin resistance [86].
Glucocorticoid produces hyperglycemia by inducing the transcription genes for hepatic gluconeogenic enzymes. Hyperglycemia in turn inhibits pancreatic beta cells, an effect that is readily overcome by a parallel event of excessive (circulating) insulin [87,88]. However the latter is invariably cytotoxic. For instance, insulin stimulation of its receptor prevents translocation of FoxO1 into the nucleus of beta cells, which in turn promotes apoptosis by the suppression of Pdx1 gene [89]. Such untoward effect may be potentiated by mitochondrial toxicity that results from concurrent use of CNI, particularly tacrolimus [90]. Ultimately, persistent stimulation of a fewer number of beta cells causes pancreatic cell hypertrophy [91].
It is pertinent to mention that in addition to risk categorization by epidemiologic profiles, assessment of genetic susceptibility are potential tools for (the) selection of patients that may benefit from individualized therapeutic approach. In this regard, genes that are associated with greater risk of developing NODAT are vitamin D receptor (VDR), hepatocyte nuclear factor (HNF1A), DUSP9 locus on X chromosome, and voltage-gated potassium channel (KCNQ1) [92,93]. Because most trials on this subject are not randomized, large scale studies will be required for the validation of this approach.
Partly due to intensity of immunosuppression, dyslipidemia occurs within the first year in more than 80% of adult renal allograft recipients [94]. Patients who had hyperlipidemia before engraftment are more likely to have a persistent disorder after transplantation [95]. Interestingly, the most significant risk factor is the cumulative dose of corticosteroid [96]. Chronic steroid use increases free fatty acid synthetase and up-regulates hepatic synthesis of very low-density lipoprotein (VLDL) [96]. It also reduces the synthesis of low-density lipoprotein (LDL) receptor and inhibits the enzymatic activity of lipoprotein lipase [97]. The sum effect is an increase in the serum total cholesterol, high serum triglyceride, and elevated level of VLDL [98,99]. However, depicting a striking similarity with HD patients, due to a concurrent reduction in Apo-A lipid fraction, elevated level of plasma high-density lipoprotein (HDL) cholesterol does not confer a cardiovascular benefit [95,100]. In addition, chronic steroid treatment potentiates the lipemic effects of both CsA and sirolimus [27,101-103]. Because CsA is transported in the circulation by LDL cholesterol particles, prednisolone enhances its intracellular uptake by LDL receptors [101].
Supporting the synergistic role of steroids, co-culture of hepatic cells in a medium containing CsA and palmitic acid causes mitochondrial dysfunction via the activation of c-Jun N-terminal kinase (JNK) [102]. In comparison, it appears tacrolimus produces less lipid disturbances [101]. Similarly, 2 weeks after adding sirolimus to the regimen of CsA and steroids, it causes a dose-dependent increase in total plasma cholesterol, LDL, triglyceride, and ApoB-100[97] [103]. Normal lipid levels are restored by about 4 weeks after discontinuation of the drug [103]. In animal model, sirolimus impairs hydrolysis of circulating triglyceride, cellular uptake of fatty acid, and lipid synthesis (lipin 1) by down-regulation of peroxisome proliferator-activated receptor-γ 2 (PPAR-γ 2) [104]. Additional mechanism of action is by the blocking of modulatory effects of mTORC1 on adipogenesis through the Akt-mediated phosphorylation of tuberous sclerosis complex 2 [104].
Chronic steroid treatment promotes thrombotic vascular disease by increasing the serum concentration of plasminogen activator inhibitor antigen (PAI-1) [105]. This relationship is demonstrated in a study that showed a significant reduction in the stimulated fibrinolytic capacity in heart transplant patients who were treated with steroid-based protocol [105]. By combining steroids with CsA, this process may be aggravated by endothelial injury that is promoted by the calcineurin inhibitor [106]. Thus experimental rat model of aortic allograft developed proliferative hyaline thickening of the vascular wall within 2 months of CsA treatment but not with the use of AZA [106]. Similarly, sirolimus may potentiate chronic steroid-induced vasculopathy [107]. It inhibits smooth muscle cell hyperplasia by up-regulating the gene for the transcription of nitric oxide synthase [108]; and reduces synthesis of vascular endothelial growth factor (VEGF) in tumor cell lines [109]. On the other hand, as previously mentioned, loading dose of steroids [during transplant surgery] may be potentially beneficial to endothelial health. It reverses tacrolimus up-regulation of pro-oxidative, asymmetric dimethyl-arginine and endothelial nitric oxide synthase [110].
The initial wave of clinical trial on steroid minimization was conducted in pediatric population out of concern for its negative impact on longitudial skeletal growth [111]. A two-year randomized trial of late steroid withdrawal (>3 months) using cyclosporine and mycophenolate mofetil as maintenance therapy showed a positive catch-up growth and a favorable metabolic impact [105,112]. Similar result was obtained in children younger than 5 years of age while improved nutritional status was observed in steroid-free pediatric recipients of intestinal transplant [106,113].
Despite normal serum calcium, phosphorous and intact PTH, osteitisfibrosa, osteomalacia, and adynamic bone disease are common findings on bone biopsy obtained from transplant recipients [114]. There is positive correlation between cumulative steroid dose, loss of bone mineral density and the incidence of pathological fracture [115]. In addition to steroid use, delayed restoration of parathyroid gland, metabolic acidosis, vitamin D deficiency, and inadequate graft function may accelerate bone disease [114]. Dexamethasone inhibits renal tubular reabsorption of calcium and/ or phosphorous by modulation of serum glucocorticoid kinase [116,117], Na+/H+ exchanger [118] and Na-Pi 4 co-transporter [119]. It decreases bone-forming osteoblastic cells, increases apoptosis of osteocytes, and attenuates both insulin-like growth factor-1 [IGF-1] and transforming growth factor-b (TGF-b) [120].
Furthermore, it stimulates osteoprotegerin ligand [OPG-L] and causes inactivation of its soluble neutralizing receptor, osteoprotegerin [OPG] [120]. Lower doses of steroids, alternate-daily regimen, and correction of hypogonadism may potentially ameliorate the severity of bone disease [121]. Indeed patients on GC withdrawal protocol sustain a net gain in BMD; a benefit that was more pronounced in patients with severe kidney disease [122]. There are limited data on the safety and effectiveness of bisphosphonates in solid organ transplantation. Its use in liver transplant recipients was associated with the preservation of trabecular bone mineralization but there was no (beneficial) therapeutic effect on the cortical bone mass [123].
We’ve achieved a remarkable stride in the provision of targeted immunosuppression against alloreactive T lymphocytes. This has reduced the attrition rate of renal allograft in the first year to the barest minimum; about 5-10%. Nevertheless inadequacy of the currently available ISA has led to increasing relevance of chronic antibody mediated injury as a potential reason for late onset graft losses. The broad-spectrum activity of steroids against B-and T- lymphocyte immune responses has played a beneficial modulatory role over the years. However, many transplant patients have paid a huge price in the form of adverse metabolic effects and CV complications. To this end, it is commendable that steroid sparing regimen have been successfully used in low risk transplant recipients. Steroid avoidance in high immunological risk recipients often necessitates [induction] treatment with potentially toxic lymphocyte depleting agents. Finally, it is very likely that steroids will continue to play a significant role in the prevention and treatment of immune mediated allograft injury. In the near future, steroids may be most useful in the context of risk categorization to identify drugs that are best suited for individual patients.
The author thank Dr Renee Gardner, Dr Fasika Tedla, Dr Costa Dimitriades, Dr Ricardo Sorensen, Dr Matti Vehaskari and Dr Susan Gottesman for their assistance with proofreading.