MEN-1, Insulinoma, Prolactinoma
Multiple endocrine neoplasia 1 (MEN1) syndrome is a rare heritable disorder characterized by neoplasia or hyperplasia of the parathyroids, enteropancreatic tissues, anterior pituitary and other tissues. Clinical diagnostic criteria for MEN 1 include presence of at least 2 of the 3 groups of endocrine tumors as mentioned above [1]. Here, we are reporting an uncommon presentation with neuropsychiatric symptoms, subsequently diagnosed with insulinoma along with additional findings consistent with MEN 1.
A twenty-one years-old Indian female patient presented with episodes of nervousness, irritability and confused behavior for a period of 20 days, particularly when a meal was delayed. About two days prior to presentation she developed an episode of loss of consciousness which recovered when she was administered dextrose containing intravenous fluids at a local clinic. She was then referred to our department for further evaluation. There was no history of chest pain, dyspnea, involuntary movements of body, frothing from mouth, tongue bite, urinary incontinence, any discharge from nipples, jaundice or facial puffiness. There was no history suggestive of factitious hypoglycemia. On physical examination, she was thinly built with a body mass index (BMI) of 22.6 Kg/m2. She was Tanner IV for pubic hair and genital development. A resting pulse rate of 114/min and blood pressure of 134/86 mm of Hg was noted. No other remarkable physical findings were present. The patient was then kept fasting in the ward and blood glucose was monitored hourly. She developed symptoms of sympathetic over activity and blood glucose was found to be ≤ 45 mg/dl and critical blood samples were drawn. She had simultaneously high serum insulin 4.65 microU/ml (to diagnose insulinoma, it should be > 3 μU/ml at the time of hypoglycemia) and a high C-peptide level of 5.0 ng/ml (to diagnose insulinoma, it should be > 0.6 ng/ml at the time of hypoglycemia) despite hypoglycemia [2]. Plasma thyroid stimulating hormone (TSH) was within normal limits. Serum cortisol was 9.18 μg/dl (normal range: 4.3-22.4 μg/dl) and growth hormone (GH) was 8.75 ng/ml (normal < 10 ng/ml in females). Glycated hemoglobin (HbA1C) was 4.33% indicating chronic hypoglycemia. Ultrasound (USG) examination of the whole abdomen revealed a hypoechoic lesion (16 × 14 mm) in the tail of the pancreas along with a small (4 mm) right renal calculus. Contrast enhanced computer tomography (CT) scan of whole abdomen showed arterial phase enhancement in the tail of the pancreas (Figure 1). The CT scan finding along with history of hypoglycemia was suggestive of a pancreatic neuro-endocrine tumor most likely an insulinoma. Evaluating the cause of renal calculus, serum calcium was ordered which was high 11.8 mg/dl (normal range 8.8-10.2 mg/dl) along with raised PTH 110.6 pg/ml (normal range 11.1-79.5 pg/ml). Serum phosphate was 2.61 mg/dl (normal range 2.5-4.5 mg/dl). USG examination of neck was done to evaluate hyperparathyroidism, which revealed a hypoechoic lesion (11 mm × 16 mm) with increased vascularity at inferior angle of thyroid s/o Parathyroid adenoma. Presence of two endocrine neoplasm warranted further investigation as possibility of MEN was considered. Serum prolactin was high (67.27 ng/ml, normal range: 1.9-25 ng/ml) and GH was normal (8.75 ng/ml, normal range < 10 ng/ml for females). Magnetic resonance imaging (MRI) scan of sella showed a small eccentric lesion in the right half of anterior pituitary measuring 2 mm × 2 mm suggestive of a microadenoma (Figure 2). The presence of three endocrine tumors i.e. enteropancreatic tumor - insulinoma, parathyroid adenoma and pituitary microadenoma (prolactinoma) established the diagnosis of MEN type 1. The prolactinoma was managed with dopamine agonist cabergoline. For insulinoma, distal pancreatectomy with splenectomy was done after Ga68 Dotatate positron emission tomography (PET) scan to rule out metastasis for insulinoma. Histopathology examination revealed tumor cells positive for synaptophysin (diffuse strong positive) and chromogranin (diffuse strong positive) with Ki-67 of 05% suggested a low grade (grade-2) neuroendocrine neoplasm. A parathyroid adenoma was detected on histopathology examination of excised parathyroid gland. She was discharged symptom free and kept on regular follow up to monitor any possible recurrence. She is currently taking dopamine agonist cabergoline and regular prolactin levels monitoring.
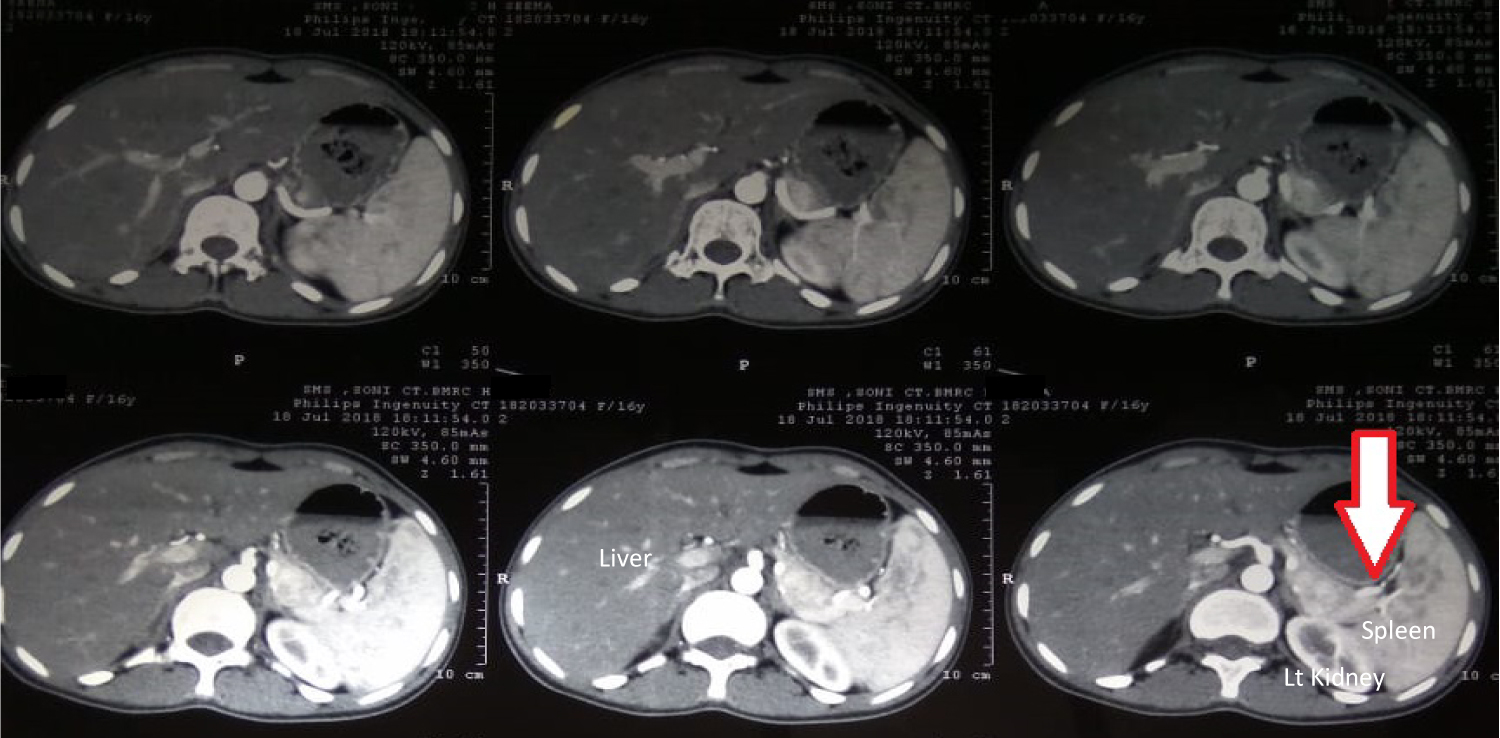 Figure 1: CECT abdomen showing arterial phase enhancement in the tail of pancreas.
View Figure 1
Figure 1: CECT abdomen showing arterial phase enhancement in the tail of pancreas.
View Figure 1
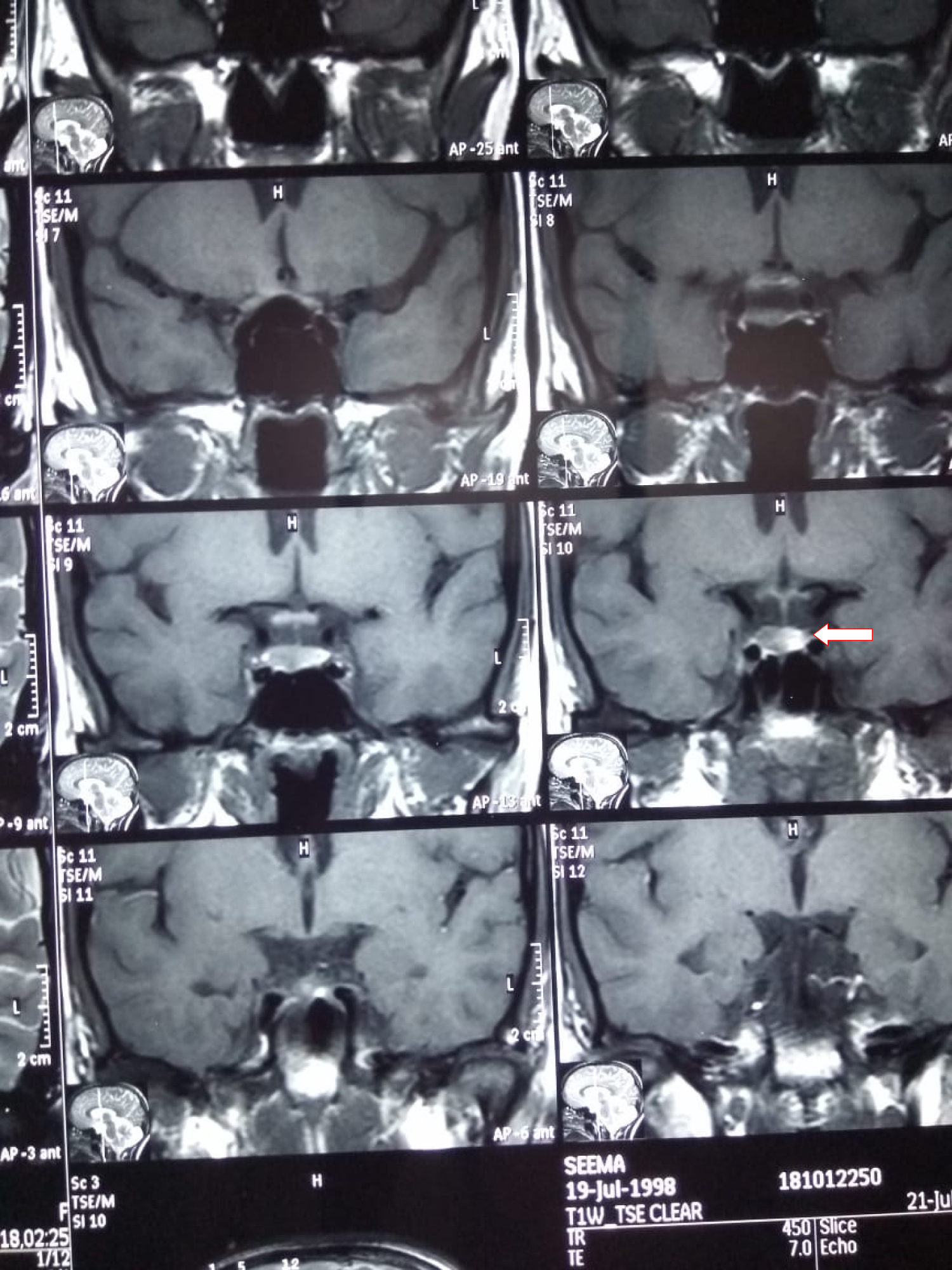 Figure 2: MRI brain with sella cuts showing anterior pituitary microadenoma.
View Figure 2
Figure 2: MRI brain with sella cuts showing anterior pituitary microadenoma.
View Figure 2
The term multiple endocrine neoplasia (MEN) encompasses various clinical inherited syndromes comprising of various benign or malignant tumors involving different endocrine glands. MEN - 1 affects 2-20 per 100,000 individuals. Three main tumor types are recognized: parathyroid adenomas, pancreatic neuroendocrine tumors (NET) and pituitary tumors. MEN 2 are classified into three clinical subtypes: (1) MEN2A: Medullary carcinoma of thyroid (MTC) associated with pheochromocytoma and hyperparathyroidism; (2) MEN2B: MTC associated with mucosal neuromas, pheochromocytoma, marfanoid body habitus, gastrointestinal ganglioneuromatosis and myelinated corneal nerves; and (3) Familial MTC: Only MTC is present. MEN2B is a more aggressive disease than MEN2A or familial MTC [3].
Various cutaneous tumors are commonly found in MEN1 but are often subclinical. These include multiple angiofibromas, collagenomas, and lipomas. In contrast to tuberous sclerosis the angiofibromas in patients with MEN1 are smaller in size and less numerous. In addition, angiofibromas in MEN1 commonly involve the upper lip and vermillion border of the lip, whereas in tuberous sclerosis, angiofibroma often spare the upper lip region. Collagenomas are multiple small, light to skin-colored, firm, round to oval papules that are 0.2-2 cm in diameter. They are mostly located on the upper part of the trunk and on the neck [4].
MEN-1 mostly are asymptomatic and often present with symptoms of hyperparathyroidism. The neuro-psychiatric symptoms due to hypoglycemia are seen very rarely. These include a wide range of autonomic responses like sweating, weakness, hunger as well as neuroglycopenia symptoms as irritability, confusion and loss of consciousness. Often, in non-diabetic persons, these manifestations are misdiagnosed as some underlying neuropsychiatric disorder. Insulinoma, a rare cause of hypoglycemia is diagnosed with insulin concentrations of at least 3 μU/ml (18 pmol/l) and c-peptide concentrations of at least 0.6 ng/ml (0.2 nmol/l) when the fasting glucose concentrations are below 55 mg/dl without any detectable oral hypoglycemic agent levels and no circulating insulin antibodies [5]. Insulinoma in MEN-1 presents in second to fourth decade of life as compared to sporadic cases which present late. MEN-1 insulinoma are often small, may be multiple and may be associated with simultaneous presence of other islet cell tumors. Other gastro-intestinal tumor in MEN-1 is gastrinoma causing Zollinger-Ellison syndrome (ZES), which is also the most common cause of symptomatic disease in MEN-1. Non-functioning pancreatic tumors may be malignant and can metastasize to liver. Gallium-Dotatate PET/CT scanning has been reported to have especially high sensitivity for detecting neuroendocrine tumors in MEN-1 [6]. A combination of various pharmacological and surgical approaches is used in treatment.
Pituitary adenomas are clinically apparent in 15 to 20 percent of patients with MEN-1. Prolactinoma is the most common type of pituitary adenoma, but somatotroph, corticotroph, gonadotroph, and clinically nonfunctioning tumors can also occur in MEN-1. Clinical manifestations of pituitary tumors in MEN-1 are similar to those with sporadic tumors, and include those that are due to hormone excess, and those that are related to the size of the tumor [6]. Patients with prolactinomas may present with amenorrhea, galactorrhea and infertility in women; or erectile dysfunction and infertility in men. Patients having excessive secretion of GH or ACTH may present with the signs or symptoms of hormone excess e.g. gigantism or acromegaly. Individuals with enlarging pituitary tumors may experience headaches and signs of compression of adjacent structures such visual field disturbances in the setting of optic chiasm compression, and/or hypopituitarism. Treatment of pituitary tumors in MEN-1 does not differ from sporadic cases and include trans-sphenoidal surgical removal of the tumor. Prolactinomas respond well to therapy with dopamine agonists, which may be used for a long period of time.
Parathyroid tumor is most common manifestation of MEN-1, causing primary hyper-parathyroidism. It has over 90 percent penetrance by age of 40 to 50 years [7]. Presence of symptomatic or marked hypercalcemia, nephrolithiasis, and evidence of bone disease such as diminished bone density or fracture are common manifestations. Similar to sporadic hyperparathyroidism, the majority of patients are asymptomatic and routine biochemical screening revealing hypercalcemia or renal calculi detected on abdominal ultrasonography guides towards accidental diagnosis of primary hyperparathyroidism. Indications for surgical intervention are similar to that of sporadic primary hyperparathyroidism as marked hypercalcemia, nephrolithiasis and evidence of bone disease as diminished bone density or fracture. Zollinger-Ellison syndrome is difficult to control with medications in patients with MEN-1 since hypercalcemia worsens hypergastrinemia, thus making it a rare indication for parathyroidectomy, since pharmacological therapy is highly successful in hypergastrinemia [6].
The neuro-psychiatric presentation seen in this case is unusual for MEN-1. Since all three defining endocrine organs, including pituitary prolactinoma, parathyroid adenoma, and pancreatic neuroendocrine tumors (insulinoma) were seen in our patient, it demonstrates a classic case of MEN-1. Appropriate laboratory workup and targeted imaging of involved endocrine organs are needed when the clinical suspicion for MEN-1 is high. Careful multidisciplinary management and follow-up is required for patients diagnosed with MEN-1.
None.
This is to state that all the authors contributed equally for the preparation of this manuscript.