Abstract
Based on the key role of hyperglycemia-caused oxidative stress in the formation of diabetic cardiomyopathy, increased antioxidant process would best restorative approach for control of diabetic cardiomyopathy. The increased nitrative stress and peroxynitrite, activation of iNOS and TGF-B1 play a crucial role in the pathogenesis of diabetic cardiomyopathy. Reduction of the expressions of the NOS-2 and TGF-B1 appears to be one of the important mechanisms for resveratrol effect on heart.
Keywords
Type 2 diabetes, Antioxidant, Hyperglycemia, Redox status, Cardiomyopathy, Resveratrol
Introduction
Coronary artery disease leading to myocardial infarction and heart failure is a chronic complication of diabetes [1]. After a myocardial ischemic event, diabetes is associated with increased adverse outcomes in terms of both morbidity and mortality over the short and long term [2].
Diabetic cardiomyopathy, a disorder of the heart muscle in diabetic patients, is one of the major causes of heart failure. Diabetes related cardiomyopathy has been defined as diabetic-mediated ventricular dysfunction independently of any vascular disease and hypertension manifesting initially by diastolic dysfunction, later by systolic dysfunction, and finally by heart failure [3]. In older studies, several mechanisms have been discussed for onset and development of diabetic cardiomyopathy, it is thinked that oxidative stress resulting from diabetes-mediated hyperglycemia, lipotoxicity and mitochondrial dysfunction plays a main role in the development of cardiomyopathy in diabetic patients [4,5].
The mechanism of diabetic cardiomyopathy are different and may related increased oxidative/nitrosative stress with induced hyperglycemia [6], and activation of its downstream effector pathways (e.g. poly(ADP-ribose) polymerase (PARP)) [7,8], changes in the composition of extracellular matrix with increased myocardial fibrosis and inflammation [9,10].
Hyperglycemia has been shown to inhibit the prosurvival effect of VEGF, leading to cell apoptosis via tyrosine nitration of PI3Kinase that results in Akt inactivation and increased p38 mitogen-activated protein kinase activation in cells [11]. NO, synthesized from L-arginine by 3 NO synthase (NOS) enzymes, is a biological mediator with multiple actions [12]. Two constitutively present enzymes are found in neuronal and endothelial cells, respectively; the third form is inducible in many cells by endotoxin and cytokines, such as interleukin-1, interferon-γ, and TNF-α [13,14].
Experimental studies have shown that induced NO synthesis has a negative inotropic effect on cardiac cells [15] and that high levels of NO produced by inducible NOS (iNOS) are cytotoxic [16,17]. Recent studies have suggested that generation of nitric oxide (NO) in the endocardium and myocardium regulates cardiac function in a paracrine and autocrine fashion [18,19]. In patients with cardiac dysfunction, can be attributed to excessive NO production resulting from cytokine-induced expression of iNOS in myocardial and vascular tissues [20,21].
The authors showed that expression of iNOS protein with a high prevalence of TNF-alpha protein expression in cardiac myocytes related with cardiomyopathy. Some researchers have previously shown that inducible NO synthase (iNOS) expression was significantly elevated in STZ diabetic rat hearts [22] and once induced, iNOS is known to generate large amounts of NO until the enzyme is degraded [23]. Some cytokines, as example IGF-I, TGF-β1, connective tissue growth factor, platelet-derived growth factor, basic fibroblast growth factor, and epidermal growth factor, have been reported to both increase cellular proliferation (intestinal smooth muscle cells or myofibroblasts) and increase synthesis of ECM or collagen [24,25].
We know that the collagen content is increased in myocardium of patients with cardiomyopathy, indicating changes in the collagenous framework of the heart [26,27]. Moreover, some authors found correlation between the myocardial collagen and fasting plasma glucose, mRNA expressions of TGF beta [28]. Also, AGEs activate several critical molecular pathways, which trigger production of profibrogenic growth factors, connective tissue growth factor (CTGF), and TGFβ1 [29,30] as well as the inflammatory response. Transforming growth factor (TGF)-β1 co-regulates MMP expression, it affects fibroblast proliferation and regulates ECM production by promoting collagen synthesis and reducing collagen degradation (Figure 1) [31,32].
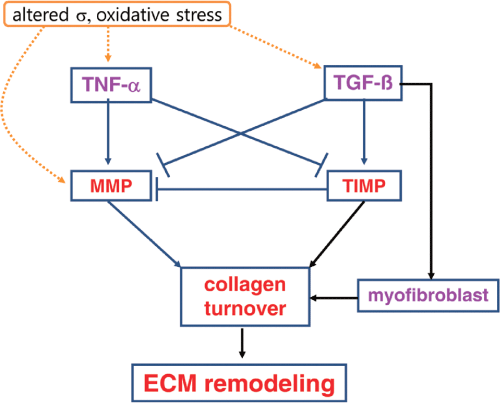 Figure 1: Collagen turnover in the heart. Changed mechanical stress (σ) and oxidative stress may trigger TNF-α, TGF-β, and MMP. TNF-α stimulated the matrix metalloproteinases (MMP) and inhibit the tissue inhibitors of metalloproteinases (TIMP). In a result, MMP degrades collagens, but TIMP and myofibroblast inhibit collagen degradation and promote collagen synthesis, finally determine collagen ECM remodeling.
View Figure 1
Figure 1: Collagen turnover in the heart. Changed mechanical stress (σ) and oxidative stress may trigger TNF-α, TGF-β, and MMP. TNF-α stimulated the matrix metalloproteinases (MMP) and inhibit the tissue inhibitors of metalloproteinases (TIMP). In a result, MMP degrades collagens, but TIMP and myofibroblast inhibit collagen degradation and promote collagen synthesis, finally determine collagen ECM remodeling.
View Figure 1
.
Aharinejad and collageous found that TGF-β1 expression is increased in myocardium of DCM patients; and that TGF-β1 stimulation in DCM cardiac tissue significantly increased collagen type I but reduced MMP-1 [33]. Another researchers were found that resveratrol can be significant upregulation of the protein expression profiles of vascular endothelial growth factor (VEGF) [34].
For many years, new agents have been tried to get away from oxidative attack cardiovascular disorders. Resveratrol (trans-3,5,4'-trihydroxystilbene), a polyphenolic compound and naturally occurring phytoalexin, has been designated the active agent [35] present in red wine. In a previous study, resveratrol reduced the oxidative stress and increased inducible nitric oxide synthase (iNOS) mRNA expression which leading to reduction of cardiomyocyte apoptosis and infarct size. In another study researchers showed that induced effect of resveratrol via VEGF, iNOS and eNOS increased expression. This situation provided significant cardioprotection as evidenced by the reduction of infarct area and increased capillary density [36] in the rats myocardial tissue. The definite mechanism for the beneficial effects of resveratrol may be due to nitric oxide (NO) production in endothelial cells, and in the heart [37]. Giovannini, et al. [37] and Naderali, et al. [38] demonstrated that upregulation of NO is a principal factor for the anti-ischemic function of resveratrol [39].
Hong and collageous showed that resveratrol suppress the protein expression of TGF-B1, TGFR-I (TGF-B type I receptor) and TGFR-II (TGF-B type II receptor) in renal epithelial cells [40]. In previosuly studies, resveratrol reduced the mRNA expression of the inflammatory mediators TNF-α and IL-1β, as well as the mRNA expression of collagen type I and TGF-β in the dimethylnitrosamine model of liver fibrosis in rats [41,42]. Diabetic rats supplemented with antioxidant showed decreased glucose, HbA1c and increased the plasma insulin levels. As β-cells in particular, are highly susceptible to oxidative stress, hyperglycemia and dyslipidemia [43-45] increase in oxidative stress in STZ diabetic rat which may induce apoptosis of β-cell.
In last a study has depicted that 8 weeks treatment with resveratrol (2.5 mg/kg/day; ip.) improves diabetic heart function through reducing ventricular inflammation and remodeling [46]. Also in the other recent study, it has been shown that resveratrol given at 2.5 mg/kg/day orally for 4 weeks prevented from cardiac and vascular dysfunction in diabetic rats through its effect on redox balance [47].
Accumulating evidence suggests that hyperglycemia induces ROS [46] and overproduction of ROS is associated with apoptosis in the diabetic heart [48]. In response to the increased oxidative stress, the antioxidant enzymes catalase which act as a defense system, are also induced to protect the cell from oxidative stress [49]. It is observed that, in case of elevated oxidative stress, cells with increased levels of antioxidants are hypersensitive to oxidative stress rather than protected from it thus rendering the cells resistant to oxidative stress.
iNOS is induced by inflammatory cytokines and produces a much higher level of NO compared with constitutive NOS [50]. Cardiac myocytes, as well as a number of other parenchymal cells within the myocardium, including the endothelium of the coronary microvasculature, endocardium, and infiltrating inflammatory cells, are all able to expression of the NOS in response to soluble inflammatory mediators [51].
That have reported fibronectin to be elevated in the kidney, heart, and retina of diabetic rats; in human mesangial cells, hyperglycemia has also been found to induce expression of TGFβ-1, which precedes accumulation of extracellular matrix [52]. In a study, resveratrol downregulated the expression of MCP-1-related proteins, including TGF-B1, TGFR-I or II related to NAD(P)H oxidase system, demonstrating that resveratrol is effective in reducing the oxidative stress induced by oxalate in renal epithelial cells via the TGF-β signaling pathway [39].
The findings of some studies indicate that inhibition of oxidative and nitrosative stress with antioxidant treatment improves LV function and minimizes apoptotic cell death in STZ-induced diabetic rat. Besides NADPH oxidase and mitochondrial electron chain as a source of ROS [53,54] in the diabetic myocardium that may play an essential role in diabetic cardiac dysfunction and cardiomyopathy.
In a result, the results of these studies demonstrate that hyperglycemia induces increased expression of NOS-2 and TGF-B1 in cardiomyocytes and that the increased cardiac muscle thickness in the diabetic heart. Reduction of the expressions of the NOS-2 and TGF-B1 appears to be one of the important mechanisms for resveratrol effect on heart.
Conflict of Interests
The authors have no conflict of interest.
References
-
Jacobs J, Sena M, Fox N (1991) The cost of hospitalization for the late complications of diabetes in the United States. Diabet Med 8: S23-S29.
-
Abbud ZA, Shindler DM, Wilson AC, Kostis JB (1995) Effect of diabetes mellitus on short- and long-term mortality rates of patients with acute myocardial infarction: A statewide study: Myocardial Infarction Data Acquisition System Study Group. Am Heart J 130: 51-58.
-
auters C, Lamblin N, Mc Fadden EP, Van Belle E, Millaire A, et al. (2003) Influence of diabetes mellitus on heart failure risk and outcome. Cardiovasc Diabetol 2: 1.
-
Bell DS (1995) Diabetic cardiomyopathy. A unique entity or a complication of coronary artery disease? Diabetes Care 18: 708-714.
-
Rodrigues B, McNeill JH (1992) The diabetic heart: Metabolic causes for the development of a cardiomyopathy. Cardiovasc Res 26: 913-922.
-
Pacher P, Szabo C (2006) Role of peroxynitrite in the pathogenesis of cardiovascular complications of diabetes. Curr Opin Pharmacol 6: 136-141.
-
Pacher P, Liaudet L, Soriano FG, Mabley JG, Szabo E, et al. (2002) The role of poly (ADP-ribose) polymerase activation in the development of myocardial and endothelial dysfunction in diabetes. Diabetes 51: 514-521.
-
Pacher P, Szabo C (2005) Role of poly (ADP-ribose) polymerase-1 activation in the pathogenesis of diabetic complications: endothelial dysfunction, as a common underlying theme. Antioxid Redox Signal 7: 1568-1580.
-
Westermann D, Rutschow S, Jager S, Linderer A, Anker S, et al. (2007) Contributions of inflammation and cardiac matrix metalloproteinase activity to cardiac failure in diabetic cardiomyopathy: The role of angiotensin type 1 receptor antagonism. Diabetes 56: 641-646.
-
Westermann D, Van Linthout S, Dhayat S, Dhayat N, Escher F, et al. (2007) Cardioprotective and anti-inflammatory effects of interleukin converting enzyme inhibition in experimental diabetic cardiomyopathy. Diabetes 56: 1834-1841.
-
El-Remessy AB, Bartoli M, Platt DH, Fulton D, Caldwell RB (2005) Oxidative stress inactivates VEGF survival signaling in retinal endothelial cells via PI-3-kinase tyrosine nitration. J Cell Sci 118: 243-252.
-
Lowenstein CJ, Hill SL, Lafond-Walker A, Wu J, Allen G, et al. (1996) Nitric oxide inhibits viral replication in murine myocarditis. J Clin Invest 97: 1837-1843.
-
Freeman GL, Colston JT, Zabalgoitia M, Chandrasekar B (1998) Contractile depression and expression of proinflammatory cytokines and iNOS in viral myocarditis. Am J Physiol 274: 249-258.
-
Habib FM, Springall DR, Davies GJ, Oakley CM, Yacoub MH, et al. (1996) Tumor necrosis factor and inducible nitric oxide synthase in dilated cardiomyopathy. Lancet 347: 1151-1155.
-
Moncada S, Palmer RM, Higgs EA (1991) Nitric oxide: Physiology, pathology and pharmacology. Pharmacol Rev 43: 109-142.
-
Finkel MS, Oddis CV, Jacob TD, Watkins SC, Hattler BG, et al. (1992) Negative inotropic effects of cytokines on the heart mediated by nitric oxide. Science 257: 387-389.
-
Kroncke KD, Fehsel K, Kolb-Bachofen V (1995) Inducible nitric oxide synthase and its product nitric oxide, a small molecule with complex biological activities. Biol Chem Hoppe-Seyler 376: 327-343.
-
Brady AJ, Poole-Wilson PA, Harding SE, Warren JB (1992) Nitric oxide production within cardiac myocytes reduced their contractility in endotoxemia. Am J Physiol 263: 1963-1966.
-
Brutsaert DL, Andries LJ (1992) The endocardial endothelium. Am J Physiol 263: 985-1002.
-
Anggard E (1994) Nitric oxide: Mediator, murderer, and medicine. Lancet 343: 1199-1206.
-
De Belder AJ, Radomski MW, Martin JF, Moncada S (1995) Nitric oxide and the pathogenesis of heart muscle disease. Eur J Clin Invest 25: 1-8.
-
Nagareddy PR, Xia Z, McNeill JH, MacLeod KM (2005) Increased expression of iNOS is associated with endothelial dysfunction and impaired pressor responsiveness in streptozotocin-induced diabetes. Am J Physiol Heart Circ Physiol 289: 2144-2152.
-
Mac Micking J, Xie QW, Nathan C (1997) Nitric oxide and macrophage function. Annu Rev Immunol 15: 323-350.
-
Burke JP, Muslow JJ, O'Keane C, Docherty NG, Watson RW, et al. (2007) Fibrogenesis in Crohn's disease. Am J Gastroenterol 102: 439-448.
-
Zimmermann EM, Sartor RB, McCall RD, Pardo M, Bender D, et al. (1993) Insulin like growth factor I and interleukin 1β messenger RNA in a rat model of granulomatous enterocolitis and hepatitis. Gastroenterology 105: 399-409.
-
Pauschinger M, Knopf D, Petschauer S, Doerner A, Poller W, et al. (1999) Dilated cardiomyopathy is associated with significant changes in collagen type I/III ratio. Circulation 99: 2750-2756.
-
Zhong M, Zhang W, Miao Y, Ma X, Gong HP, et al. (2006) The role of glucose/TSP-1/TGFbeta1 signal pathways in diabetic cardiomyopathy. Zhonghua Xin Xue Guan Bing Za Zhi 34: 217-221.
-
Candido R, Forbes JM, Thomas MC, Thallas V, Dean RG, et al. (2003) A breaker of advanced glycation end products attenuates diabetes induced myocardial structural changes. Circ Res 92: 785-792.
-
Zhou G, Li C, Cai L (2004) Advanced glycation end-products induce connective tissue growth factor-mediated renal fibrosis predominantly through transforming growth factor β-independent pathway. Am J Pathol 165: 2033-2043.
-
Nishikawa T, Araki E (2007) Impact of mitochondrial ROS production in the pathogenesis of diabetes mellitus and its complications. Antioxid Redox Signal 9: 343-353.
-
Aharinejad S, Krenn K, Paulus P, Schäfer R, Zuckermann A, et al. (2005) Differential role of TGF-beta1/bFGF and ET-1 in graft fibrosis in heart failure patients. Am J Transplant 5: 2185-2192.
-
Hyo-Bum Kwak (2013) Aging, exercise, and extracellular matrix in the heart. J Exerc Rehabil 9: 338-347.
-
Kopp P (1998) Resveratrol, a phytoestrogen found in red wine. A possible explanation for the conundrum of the 'French paradox'? Eur J Endocrinol 138: 619-620.
-
Fukuda S, Kaga S, Zhan L, Bagchi D, Bertelli A, et al. (2006) Beneficial effect of pharmacological intervention by resveratrol induces myocardial angiogenesis by upregulating VEGF and Tyrosine Kinase Receptor, Flk-1/KDR. Cell Biochem Biophys 44: 43-49.
-
Jager U, Nguyen-Duong H (1999) Relaxant effect of trans-resveratrol on isolated porcine coronary arteries. Arzneimittelforschung 49: 207-211.
-
Hattori R, Otani H, Maulik N, Das DK (2002) Pharmacological preconditioning with resveratrol: Role of nitric oxide. Am J Physiol Heart Circ Physiol 282: 1988-1995.
-
Giovannini L, Migliori M, Longoni BM, Das DK, Bertelli AA, et al. (2001) Resveratrol, a polyphenol found in wine, reduces ischemia reperfusion injury in rat kidneys. J Cardiovasc Pharmacol 37: 262-270.
-
Naderali EK, Doyle PJ, Williams G (2000) Resveratrol induces vasorelaxation of mesenteric and uterine arteries from female guinea-pigs. Clin Sci (Lond) 98: 537-543.
-
Hong SH, Lee HJ, Sohn EJ, Ko HS, Shim BS, et al. (2013) Anti-nephrolithic potential of resveratrol via inhibition of ROS, MCP-1, hyaluronan and osteopontin in vitro and in vivo. Pharmacol Rep 65: 970-979.
-
Hong SW, Jung KH, Zheng HM, Lee HS, Suh JK, et al. (2010) The protective effect of resveratrol on dimethylnitrosamine-induced liver fibrosis in rats. Arch Pharm Res 33: 601-609.
-
Lee ES, Shin MO, Yoon S, Moon JO (2010) Resveratrol inhibits dimethylnitrosamine-induced hepatic fibrosis in rats. Arch Pharm Res 33: 925-932.
-
Cnop M, Hannaert JC, Grupping AY, Pipeleers DG (2002) Low density lipoprotein can cause death of islet beta-cells by its cellular uptake and oxidative modification. Endocrinology 143: 3449-3453.
-
El-Assaad W, Buteau J, Peyot ML, Nolan C, Roduit R, et al. (2003) Saturated fatty acids synergize with elevated glucose to cause pancreatic beta-cell death. Endocrinology 144: 4154-4163.
-
Delucchi F, Berni R, Frati C, Stefano Cavalli, Gallia Graiani, et al. (2012) Resveratrol treatment reduces cardiac progenitor cell dysfunction and prevents morpho-functional ventricular remodeling in type-1 diabetic rats. PLoS One 7: e39836.
-
Turan B, Tuncay E, Vassort G (2012) Resveratrol and diabetic cardiac function: Focus on recent in vitro and in vivo studies. J Bioenerg Biomembr 44: 281-296.
-
Fiordaliso F, Bianchi R, Staszewsky L, Cuccovillo I, Doni M, et al. (2004) Antioxidant treatment attenuates hyperglycemia-induced cardiomyocyte death in rats. J Mol Cell Cardiol 37: 959-968.
-
Modesti A, Bertolozzi I, Gamberi T, Marchetta M, Lumachi C, et al. (2005) Hyperglycemia activates JAK2 signaling pathway in human failing myocytes via angiotensin II-mediated oxidative stress. Diabetes 54: 394-401.
-
Cai L, Wang Y, Zhou G, Chen T, Song Y, et al. (2006) Attenuation by metallothionein of early cardiac cell death via suppression of mitochondrial oxidative stress results in a prevention of diabetic cardiomyopathy. J Am Coll Cardiol 48: 1688-1697.
-
Guo Z, Xia Z, Jiang J, McNeill JH (2007) Downregulation of NADPH oxidase, antioxidant enzymes, and inflammatory markers in the heart of streptozotocin-induced diabetic rats by N-acetyl-L-cysteine. Am J Physiol Heart Circ Physiol 292: 1728-1736.
-
Csiszar A, Labinskyy N, Podlutsky A, Kaminski PM, Wolin MS, et al. (2008) Vasoprotective effects of resveratrol and SIRT1: Attenuation of cigarette smoke-induced oxidative stress and proinflammatory phenotypic alterations. Am J Physiol Heart Circ Physiol 294: 2721-2735.
-
Arstall MA, Sawyer DB, Fukazawa R, Kelly RA (1999) Cytokine-mediated apoptosis in cardiac myocytes: The role of inducible nitric oxide synthase induction and peroxynitrite generation. Circ Res 85: 829-840.
-
Murphy M, Godson C, Cannon S, Kato S, Mackenzie HS, et al. (1999) Suppression subtractive hybridization identifies high glucose levels as a stimulus for expression of connective tissue growth factor and other genes in human mesangial cells. J Biol Chem 274: 5830-5834.
-
Privratsky JR, Wold LE, Sowers JR, Quinn MT, Ren J (2003) AT1 blockade prevents glucose-induced cardiac dysfunction in ventricular myocytes: role of the AT1 receptor and NADPH oxidase. Hypertension 42: 206-212.
-
Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, et al. (2007) Mitochondrial energetics in the heart in obesity-related diabetes: Direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes 56: 2457-2466.