Congenital heart disease is the most frequent malformation. In the most recent data has a prevalence of thirteen cases per 1,000 children and six cases per 1,000 adults. Great strides in pediatric cardiovascular surgery have increased the number of survivors, especially in adulthood. Nowadays more adults are living with congenital heart disease than children, particularly in developed countries, and more than two thirds fall into the categories of moderate and high complexity defects. This growing population called: Grown up congenital heart, requires super-specialized healthcare, the profile of the new cardiovascular patient differs from the traditional adult with heart disease, the first paradigm that should be broken is to understand that cardiovascular surgery is restorative (it substantially improves the hemodynamic condition), but is not 100% curative. Surgical and survival successes have turned the pediatric patient with congenital heart disease into an adult postoperative patient with residua, sequela and complications. The adult's postoperative heart is not a natural heart, it is a transformed, "neo-formed", heart.
They will have relapses and hospitalizations due decompensation to congenital heart disease. A new specialist profile is necessary. Training in adult congenital heart disease is valid beginning from both a pediatric and an adult cardiology environment. The special needs of this population are very costly for health systems. Recognize what are the main causes of decompensation is mandatory and finally adequate care decreases prolonged hospital stays and health system cost.
Congenital heart disease, Adult patient, Grown up congenital heart
Historically, 1 out of every 100 live births has been known to have congenital heart disease (CHD). In the Canadian province of Quebec (where every individual is assigned a unique medicare number in the first year of life used to track all diagnoses and health services rendered and systematically recorded until death), Marelli, et al. performed a longitudinal study over 27 years with all individuals born between 1983 and 2010. They showed that the true prevalence of CHD in 2010 was 13.11/1,000 (12.43, 13.81) in children and 6.12/1,000 (5.69, 6.57) in adults. The prevalence increased by 11% in children and 57% in adults from 2000-2010. The prevalence in the severe CHD subgroup increased by 19% (17%, 21%) in children and 55% (51%, 62%) in adults [1]. These analyses have been extrapolated to other countries and regions of the world, showing similar results.
Great strides in pediatric cardiovascular surgery and new hybrid treatment techniques for serious CHDs have increased the number of survivors. Currently, more than two thirds of all adults with congenital heart disease (ACHDs) fall into the categories of moderate and high complexity defects, and are a real diagnostic and treatment challenge for specialists. In 1973, Joseph K Perloff had already described the changing population of congenital heart disease. During the 70s, 3,718,000 births were recorded in the United States (US), of which 28,000 had CHD (0.8%), but only 50% were able to survive beyond their first year of life. The currently accepted survival rate is more than 90% for most CHDs. In the US, particularly, from 1940-1959, survival at one year of age for high, medium and low complexity defects was 20%, 60% and 95%, respectively; likewise, from 1980-1989, survival was 85%, 90% and 95%, respectively [2]. These data were shown as the changing profile of congenital heart disease in adult life (Task Force 1 of the American College of Cardiology) [3]. On this occasion, it was shown that for the first time in history there would be more adults than children with CHD, with a 5-6% annual rate of growth, and that in 2005 there would be more than one million ACHDs. That is precisely what happened; in 2010, the US recorded 1.4 million ACHDs vs. 1 million children with CHD [4]. The current estimate is that there are 3,000 ACHDs per million inhabitants [5]. Extrapolating these results, it is estimated that there are approximately 261,000 ACHDs in Central America, and almost 1.2 million in South America. Furthermore, the European scene shows a population of 2.3 million adults with CHD vs. 1.9 million children [6].
Sixty percent of CHDs are diagnosed and treated in the first year of life, and 30% throughout childhood and adolescence up to 16 years of age. It is estimated that 10% are diagnosed in adulthood [7].
The current ACHD population is derived from two groups:
1. Newly diagnosed or never diagnosed ACHDs:
These are individuals who have survived beyond 18 years of age, and who for some reason (lack of access to healthcare, absence of major symptoms, little affectation of the functional class, or simple CHDs, among many other explanations) were never diagnosed or treated in childhood or adolescence. They have had a natural survival. This population only makes up 10% of cases, and they are mostly simple or medium complexity CHDs, according to the 2001 Bethesda classification [8]. However, there are cases of complex CHDs diagnosed and successfully treated in adulthood [9].
This type of population has primary or native CHD (without intervention) with a very different clinical and pathophysiological behavior from that of the child and adolescent. From its origin in the fetus, through childhood, adolescence and adulthood, each CHD has a very particular and unique evolution. Defects considered to have little hemodynamic repercussion in childhood (nonobstructive bicuspid aortic valve) naturally evolve to deterioration of various related structures in the adult (bicuspid aortic valve and aneurysm or aortic dissection in the adult) (Figure 1). Defects which never caused repercussions or complications in childhood will do so in adulthood (small or medium sized atrial septal defects will be complicated by arrhythmias, heart failure, and ictus, among others). The hemodynamic consequences which develop will produce changes in the primary heart anatomy (chamber dilation and valvular insufficiency, among others). These patients are a great challenge for the clinical cardiologist and echocardiographer who must start from scratch in the sequential diagnostic anatomical analysis, and once a definitive diagnosis is reached, must propose a treatment plan and repair of the congenital defect, when possible [10].
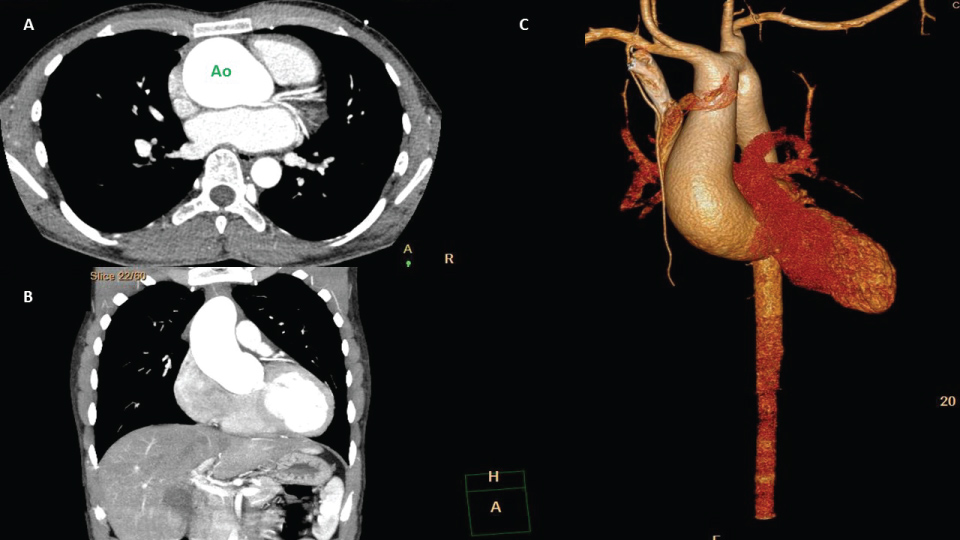 Figure 1: A,B) Ascending aortic aneurysm (transverse and coronal plane respectively); C) Ascending aortic aneurysm 3D reconstruction, in male 21-year-old. He has bicuspid aortic valve. Ao: Aorta.
View Figure 1
Figure 1: A,B) Ascending aortic aneurysm (transverse and coronal plane respectively); C) Ascending aortic aneurysm 3D reconstruction, in male 21-year-old. He has bicuspid aortic valve. Ao: Aorta.
View Figure 1
2. ACHDs who underwent surgery in childhood or adolescence:
These make up 90% of cases encountered by adult cardiologists. While the medical/surgical history of most of these individuals is known, which is a great help in determining the approach to these patients, their clinical and diagnostic focus will always be very different from that of an adult with non-congenital heart disease. The majority of these patients (85%) have medium and high complexity CHD on the 2001 Bethesda classification, and this is the group which will grow the most over the next 10-20 years.
Surgical and survival successes have turned the pediatric patient with CHD into an adult postoperative CHD patient. The adult's postoperative heart is not a natural heart, it is a transformed, ¨neo-formed¨, heart. Its original, or native, anatomy, has been modified with synthetic CHD repair elements (patches, valves, intra or extra-cardiac tubes, stents) which modify not only its primary anatomy but its whole physiology. It is no coincidence that terms such as neo-aorta and neo-pulmonary must be employed in the surgical and echocardiographic anatomical description of heart studies in patients who have undergone surgery for classic transposition of the great arteries or D-transposition (TGA) with arterial switch (referring to the aorta and pulmonary artery, respectively, since in their original anatomy they were not in the normal spatial or anatomical relationship). Likewise, in postoperative truncus arteriosus patients, where the new aortic valve (neo-aorta) has been "neo-formed" from a single trunk valve, with a different anatomical and functional placement from that of the natural aortic valve.
To complicate the picture a bit more, the same congenital defect may be repaired using different surgical techniques, which are chosen according to whether or not certain conditions associated with the original CHD are present. For instance, TGA with coronary circulation abnormalities is repaired using an atrial switch with the Senning or Mustard technique (intra-atrial channels or baffles made from autologous and synthetic materials, respectively), creating an intentional atrioventricular discordance in order to correct the circulation of the great arteries. The neo-atria are now anatomically and functionally different, and must be specifically studied, looking for complications that will develop in the future (Figure 2). When TGA is associated with a ventricular septal defect and pulmonary valve stenosis, it will be repaired using the Rastelli technique (Figure 3) (closure of the ventricular septal defect with a patch, redirection of the systemic ventricle's flow to the aorta, and placement of an extra-cardiac valved conduit between the venous right ventricle and the pulmonary branches). The Nikaidoh technique has also been used (explantation of the aortic root with a cone-shaped segment of infundibular muscle, dissection and liberation of the coronary arteries without splitting them from the trunk, sectioning of the stenotic pulmonary artery at the annulus, enlargement of the ventricular septal defect and widening of the RV outflow tract, closure of the ventricular septal defect and translocation of the aortic root towards the LV and reconstruction of the RV outflow tract with a homograft orvalved or Contegra conduit). This somewhat complex technique is also used to repair double outlet right ventricle with pulmonary stenosis.
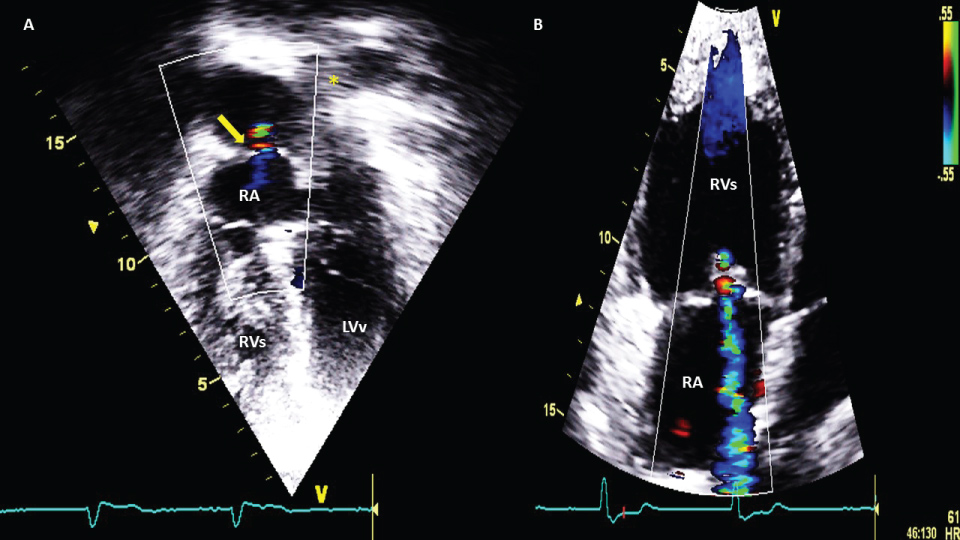 Figure 2: A) apical 4 chamber view, yellow arrow shows leak between pulmonary veins bafle and right atrium (shunt systemic to venous). RA: right atrium; RVs: right morfologic ventricle and systemic functionally, because it's conected with aorta; LVv: left morfologic ventricle and subpulmonary or venous, bucause it's conected with pulmonar artery; B) Apical 4 chamber view, tricuspid systemic insuficience is common in natural evolution due to RVs disfunction.
View Figure 2
Figure 2: A) apical 4 chamber view, yellow arrow shows leak between pulmonary veins bafle and right atrium (shunt systemic to venous). RA: right atrium; RVs: right morfologic ventricle and systemic functionally, because it's conected with aorta; LVv: left morfologic ventricle and subpulmonary or venous, bucause it's conected with pulmonar artery; B) Apical 4 chamber view, tricuspid systemic insuficience is common in natural evolution due to RVs disfunction.
View Figure 2
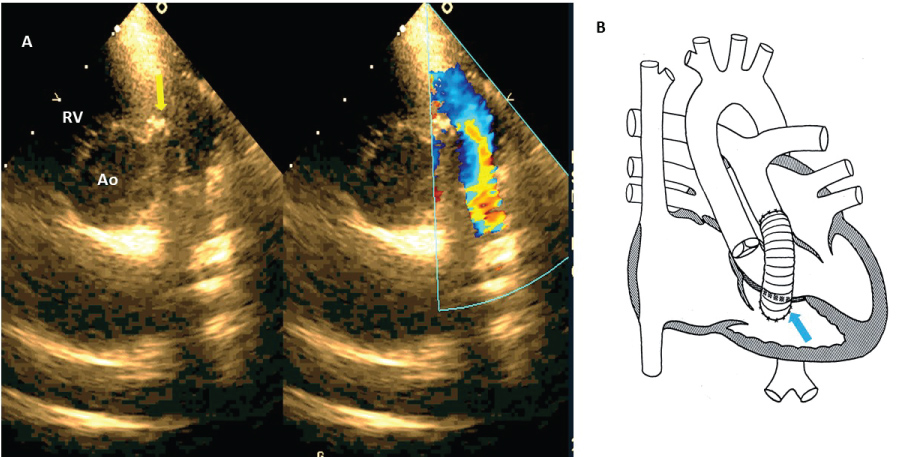 Figure 3: A) Short axis view, yellow arrow shows outflow RV conection with extracardial tube to pulmonary branches (Rastelli technique), comparative color image shows flow into extracardiac tube; B) Rastelli diagram, blue arrow shows the same in A.
View Figure 3
Figure 3: A) Short axis view, yellow arrow shows outflow RV conection with extracardial tube to pulmonary branches (Rastelli technique), comparative color image shows flow into extracardiac tube; B) Rastelli diagram, blue arrow shows the same in A.
View Figure 3
Cardiologists who are experts in ACHD not only thoroughly understand the native heart disease, along with its natural evolution in childhood and adulthood, but also understand, and should keep up to date on, a wide variety of complex surgical techniques used for repairing CHD. These surgical repairs modify the primary anatomy and physiology. With natural evolution, they develop degenerative changes which lead to deteriorated cardiac function; a function which in many cases is already impaired in severe CHD.
In 1997, Jane Somerville (British Cardiologist at Imperial College, London) described a new ACHD population who required super-specialized healthcare, especially those having undergone surgery for complex defects in childhood, or who never had surgery. She called this type of population GUCH (Grown Up Congenital Heart). Grown Up Congenital Heart patients should be centralized, and they require solutions to healthcare transition problems, knowledge of when to investigate each anomaly, experience in catheterization and cardiac interventions, heart surgeons with experience in pediatric and adult CHDs, healthcare for high risk pregnancy, life problem counseling, and a few dedicated centers with cardiologists and surgeons trained in this subspecialty [11].
Adults with congenital heart disease should be examined in non-pediatric settings. It is recommended that they be concentrated in adult congenital heart disease units (ACHDUs) with the necessary infrastructure and sufficient technological resources, and especially duly trained and skilled personnel. Recommendations for the organization of healthcare for ACHDs and subspecialty training were issued in 2014 by the Working Group on Grown-Up Congenital Heart Disease of the European Society of Cardiology (ESC) [12]:
✓ Adult/pediatric cardiologist with GUCH certification, at least 2
✓ GUCH imaging specialist (echocardiography, cardiac resonance, tomography), at least 2
✓ Congenital interventional cardiologist, at least 2
✓ CHD surgeon, at least 2
✓ Anesthesiologist with CC experience, at least 2
✓ Invasive electrophysiologist with GUCH experience, at least 1
✓ Psychologist, at least 1
✓ Social worker, at least 1
✓ Cardiovascular pathologist, at least 1
The ACHDU should provide healthcare while maintaining a close relationship with the pediatric cardiology unit and/or a cardiovascular surgery service with an active CHD program. It should treat a critical mass of patients and carry out a minimum number of procedures to be effective and maintain quality and an adequate training level.
✓ Electrocardiogram, continuous electrocardiographic monitoring (Holter), ambulatory blood pressure or event monitoring, ergometry and cardiopulmonary stress test, echocardiography, conventional radiology, computerized axial tomography (CT) and magnetic resonance (MR).
✓ Diagnostic and treatment catheterization laboratory.
✓ Diagnostic and interventional (mapping systems) electrophysiology laboratory, pacemaker/defibrillator implantation and follow-up.
✓ CHD heart surgery program.
Currently, specialized ACHD training is valid beginning from both a pediatric and an adult cardiology environment. Pediatric cardiologists have the knowledge of CHD, are familiar with the development of the heart and the behavior of CHD in childhood, and some have training in fetal cardiology and study techniques. However, when adulthood is reached, the pathophysiological disorders are very different from those of the child; some of the treatment options normally used in children are obsolete and unnecessary in adults and may even be harmful. The inherent changes in the adult heart's physiology make the functional interpretations of the heart confusing for a pediatric cardiologist unfamiliar with the assessment of adults. Adults with congenital heart disease are mostly postoperative patients with anatomic and functional changes not only due to the prior surgical repair, but also due to the natural disease progression and to diseases acquired in adulthood. Disregard for the changes and evolution of an ACHD causes treatment errors with serious consequences.
Likewise, adult cardiologists understand the adult semiological approach, anatomy, physiology, study interpretation, cardiovascular treatment and diseases. However, CHDs are a disease spectrum with which they are not familiar; learning regarding the origin of all the CHDs is very limited in their training. In most developing countries, cardiology training programs send their residents to a short (less than four months) training in CHD; they are typically sent to pediatric cardiology services, seeking to learn whatever they can in that short period of time. In Europe and North America, the opposite occurs. Pediatric cardiologists are now being sent to learn basic ACHD concepts in the ACHDUs. For adult cardiologists, the confusing CHD terminology, pathophysiology and treatment, especially in complex CHDs puts them at a great disadvantage compared to pediatric cardiologists. They practically have to begin their training from scratch.
In summary, pediatric and adult cardiologists who are to be trained in ACHD must be reeducated beginning with the anatomical and physiological bases. Pediatric cardiologists must learn adult semiology and clinical history taking, and learn to study the adult congenital heart. Adult cardiologists, in turn, must learn new CHD terminology, surgical repair techniques, the natural evolution of a great variety of CHDs, the meaning and interpretation of echocardiographic studies, and new cardiovascular treatments.
There are three levels of training, in which the objectives are:
Level 1: Basic training in problems affecting CHD patients; this enables the individual to make decisions regarding when to refer patients to specialized healthcare, not to provide care for moderately or highly complex patients.
Level 2: Additional training in ACDH to acquire experience in managing the whole spectrum of patients. The training ranges from six months to one year.
Level 3: A specialist completely trained in ACHD, with the necessary degree of knowledge to take on commitments in clinical and academic research. Requires at least two years of training.
Levels 2 and 3 can only be obtained at a center with an ACDHU.
The first paradigm that should be broken is to understand that cardiovascular surgery is restorative (it substantially improves the hemodynamic condition), but is not 100% curative, except for a few simple CHD exceptions.
With a repaired ACHD, we must answer three questions:
1. ¿ is there residual congenital heart disease?
2. ¿ what hemodynamic sequela does it or will it have?
3. ¿ what complications does or could it have?
1. Residual: According to the Oxford dictionary, it is what ¨remains, what is left¨. These are lesions deliberately left at the time of CHD repair. With few exceptions, they are obligatory defects (small residual ventricular septal defect, left atrio-ventricular valve cleft following complete AV canal repair; obligatory atrial septal defect in the case of CHD repair with pulmonary hypertension).
2. Sequelae: What ¨arises or follows¨ from a prior event; these are new cardiovascular disorders produced after carrying out CHD repair. They are lesions which are inherent to the repair, and are represented by disorders intentionally incurred during surgery (pulmonary regurgitation following Tetralogy of Fallot repair).
3. Complications: Undesired cardiovascular or systemic disorders resulting as a consequence of the therapeutic procedures, or which appear during the course of the natural evolution of the CHD (complete AV block, infectious endocarditis and ictus, among others).
In order of frequency, they are: Arrhythmias, heart failure (HF), syncope, endocarditis and pulmonary arterial hypertension (PAH). Arrhythmias make up 90% of the consults and hospitalization. By frequency, they are atrial fibrillation and flutter in 80% of cases, ventricular tachycardia (VT) 5-7%, symptomatic bradycardia 3-5%, and pacemaker malfunction 5% [13]. The causes are multifactorial and occur in both repaired and unrepaired CHD. The cavity changes due to dilation, hypertrophy and scars from previous surgeries are the anatomical substrates for their inception. Likewise, several CHDs are associated with anomalous pathways, such as Ebstein's anomaly; up to 25% of cases have accessory pathways; and CHDs such as levoisomerism present complete AV block in their development. Palliative procedures such as Rashkind's septostomy cause sinus node lesions, either directly due to tissue tearing or through injury of its irrigating artery. Fontan surgery causes right atrial dilation and sinus node dysfunction in the adult. Extensive repairs which include ventriculotomy (Tetralogy of Fallot repair) are causes of ventricular arrhythmias (VT).
These sequelae and complications which significantly deteriorate the function of the congenital heart (which is already deteriorated in many complex defect cases) should always be considered in surgical CHD repairs. Some are associated with sudden death, especially in ACHD survivors of unrepaired defects who have PAH. In Eisenmenger's syndrome, the development of myocardial fibrosis is closely related to the generation of ectopic arrhythmogenic foci and sudden death [14-16]. The presence of QRS fragmentation in complex CHDs is a factor clearly associated with sudden death [17].
Heart failure occurs as a consequence of a structural and functional heart flaw, which is incapable of maintaining an adequate cardiac output for the metabolic and hemodynamic demands. The origin of HF is multifactorial in both repaired and unrepaired hearts. Intrinsic CHD lesions such as persistent valvular stenoses, valvular regurgitation, unrepaired septal defects, chronic hypoxemia, and wide surgical repair scars cause myocardial wall remodeling and systolic and diastolic dysfunction. To further complicate matters, there are CHDs associated with cardiac muscle defects (non-compacted myocardium, Ebstein's anomaly). In complex CHDs where the RV assumes the function of the systemic ventricle (TGA repaired with an atrial switch, congenitally corrected transposition of the great arteries (CCTGA), single ventricle with right morphology) the functional deterioration occurs sooner. Another important point to keep in mind is that medical treatment alternatives for managing HF are mostly extrapolated from the management of non-congenital hearts.
Many of the methods used to treat HF in non-congenital hearts may not be adequate in congenital hearts [18]. In 2016, the Working Group of Grown-Up Congenital Heart Disease and the Heart Failure Association of the European Society of Cardiology established the first recommendations for the diagnosis and management of HF in CHD [19]. In America, the American Heart Association has issued the recommendations for the management of chronic HF in CHD [20].
A frequent problem is how to correctly classify the functional class of ACHDs in the presence of HF. Classifications such as the New York Heart Association (NYHA) categories or the modified Ross classification may underestimate the severity of disease, particularly in patients with complex or cyanotic CHD [21]. Many ACHDs are already accustomed to the physical limitations of CHD. Many think that it is normal to feel less strength, symptoms such as fatigue or palpitations are attributed to the CHD itself, and the real new HF signs and symptoms are masked by the underlying CHD. A more objective assessment with diagnostic studies such as oxygen consumption during a stress test, 3D echocardiography (Figure 4) or functional MR, which measure the ejection fraction more precisely, is a more adequate and recommended method for measuring ACHDs' functional capacity [22-25].
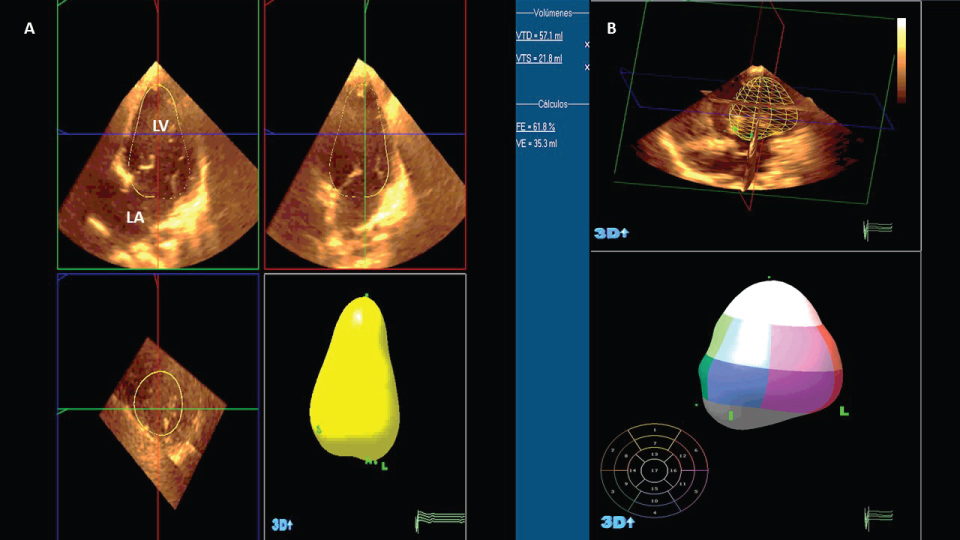 Figure 4: A) Apical 4 chamber view in classic tricuspid atresia, autoamatic 3D volumetric reconstrucction; B) Left ventricular eyection fraction (62%), automatic 3D volumetric reconstruction.
View Figure 4
Figure 4: A) Apical 4 chamber view in classic tricuspid atresia, autoamatic 3D volumetric reconstrucction; B) Left ventricular eyection fraction (62%), automatic 3D volumetric reconstruction.
View Figure 4
The growing population of ACHDs has increased the number of HF hospitalizations over the last decade. In complex CHDS, chronic HF produces 42% of the causes of death. The Dutch national registry, 'CONCOR', showed that the incidence of a first hospitalization for HF was 1.2 for every 1,000 admitted patients, increasing mortality five times compared to those not admitted. Of 24,865 patients admitted for HF, the mortality per year was 2.8%. Chronic HF caused 26% of deaths, and 19% of sudden deaths. The mean age of death due to chronic HF was 51.0 years (range: 20.3-91.2 years) [26]. In the U.S., 20% of admissions for CHD are due to uncompensated HF: Univentricular heart (Fontan) in 40%, CCTGA in 32%, and TGA repaired with atrial switch in 22% of cases.
Syncope occurs for multiple reasons (sustained arrhythmias, low output, HF, pacemaker malfunction, pulmonary thromboembolic events, pulmonary hemorrhage). Infectious endocarditis is more frequent compared to the general population (11/1,000 vs. 1.5-6/1,000 cases, respectively). The risk increases in complex CHD cases, intracavitary device bearers (pacemakers, valves) [27], associated comorbidities (immunity defects in DiGeorge syndrome), residual lesions, surgical re-interventions and prior endocarditis.
Out of the whole ACHD population, it is estimated that up to 10% may develop PAH. The 2013 international Nice classification places CHD within group 1, and there is clinical classification within this group [28] (Table 1).
Table 1: Clinical classification of pulmonary arterial hypertension associated with congenital heart disease. View Table 1
In unrepaired defects, the development of PAH depends on three factors (Figure 5):
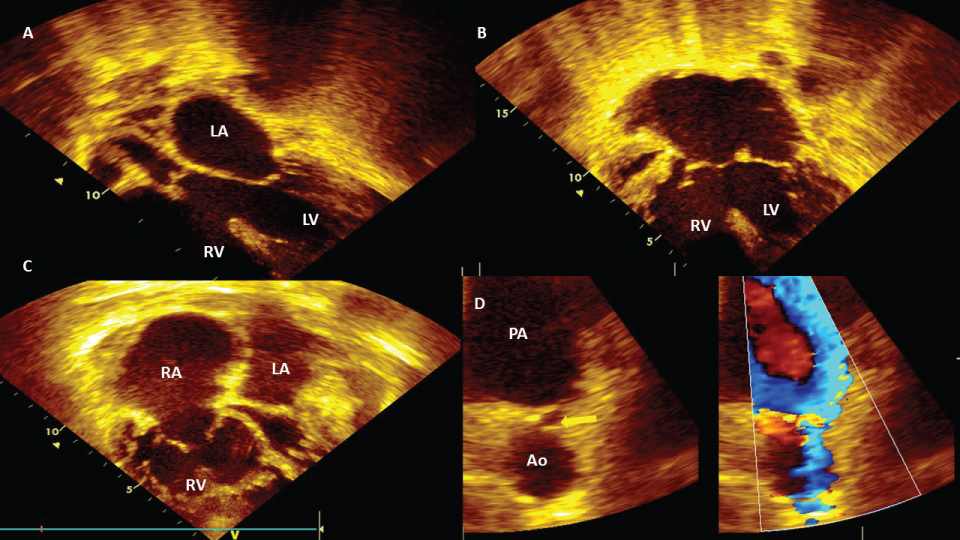 Figure 5: Post-tricuspid defects: A) Apical modificate 4 chamber view, subaortic interventricular septal defect and Eisenmenger syndrome in female 20-year-old. LV: left ventricle, LA: left atrium; B) Apical 4 chamber view, inlet septal interventricular and interatrial defect; hipertrofic RV due to pulmonary hipertensión and Eisenmenger syndrome in male 27-year-old; C) Apical 4 chamber view, dilatated RA, dilatated and hypertrofic RV, due to pulmonary hypertension; D) The same case (C), modificated short axis view with zoom in pulmonary left branche, yellow arrow shows patent ductus arteriosus, comparative color imagen shows right to left flow (venous-arterial shunt) due pulmonary hypertension and Eisenemger syndrome in female 56-year-old. PA: pulmonary artery.
View Figure 5
Figure 5: Post-tricuspid defects: A) Apical modificate 4 chamber view, subaortic interventricular septal defect and Eisenmenger syndrome in female 20-year-old. LV: left ventricle, LA: left atrium; B) Apical 4 chamber view, inlet septal interventricular and interatrial defect; hipertrofic RV due to pulmonary hipertensión and Eisenmenger syndrome in male 27-year-old; C) Apical 4 chamber view, dilatated RA, dilatated and hypertrofic RV, due to pulmonary hypertension; D) The same case (C), modificated short axis view with zoom in pulmonary left branche, yellow arrow shows patent ductus arteriosus, comparative color imagen shows right to left flow (venous-arterial shunt) due pulmonary hypertension and Eisenemger syndrome in female 56-year-old. PA: pulmonary artery.
View Figure 5
1. The size of the defect
2. Location:
• Pre-tricuspid
• Post-tricuspid
3. Time elapsed
The most extreme form of PAH is Eisenmenger syndrome (ES), commonly manifested in chronic hypoxemia with multiple systemic complications (Table 2) [29]. In Western countries It is estimated that the prevalence of PAH associated with CHD unrepaired varies between 1.6 and 12.5 cases per million adults, with 25-50% of this population affected by the ES. The chronic flow and increase of persistent pressure of the pulmonary vasculature due to an uncorrected cardiac defect with shunt left to right, develops increased pulmonary pressure and obstructive pulmonary artery disease, which leads to an increase in pulmonary vascular resistance and when it overcomes systemic resistance, it will cause a reversal of the derivation. It is estimated that 10% of patients with VSD of any size who are older than 2 years can develop ES, compared with 4-6% of patients with atrial septal defect (ASD). Of patients with large defects, all of those with truncus arteriosus, 50% of those with VSD and 10% of those with ASD will develop PAH. Of patients with ASD, the incidence of PAH is greater in those with sinus venosus defects (16%) compared with those with ostium secundum defects (4%) [30].
Table 2: Systemic consequences due to chronic hipoxemia. View Table 2
Finally, half of patients with large defects, patent ductus arteriosus or ventricular septal defects who develop ES have an earlier onset (80% during infancy) than patients with atrial septal defects (90% during adulthood) [31].
In summary, ACHDs are a constantly growing population due to the success of pediatric cardiovascular surgery, improved health care and childhood survival. The profile of the new cardiovascular patient differs from the traditional adult with heart disease, leaving ischemic heart disease, arterial hypertension, HF, acquired valve diseases and myocardial diseases, among others, to the care of general adult cardiologists. The new ACHD population needs a new duly educated and trained specialist from either the pediatric or adult field. The new ACHD specialty should provide all the necessary care, interacting with other medical specialties (internal medicine, gynecology, pulmonology, cardiovascular surgery, and anesthesiology, among others), and emphasizing multidisciplinary work which permits the prompt detection of possible complications of repaired and unrepaired CHDs. The special needs of this population are very costly for health systems. Maintaining this population's care decreases prolonged hospital stays and health system cost overruns.
To my wife and my daughters D. Lynn and Blanquita. Thanks for your support.