Adrenocortical tumors are rare neoplasms with poor prognosis and with an incidence of one in one million population. They are categorized as either functional (hormone-secreting) or silent and as either benign or malignant. We are reporting a very rare case of huge (16 cm) adrenocortical carcinoma in a 19-years-old male patient who presented with a progressively increasing right abdominal mass and uncontrolled systemic blood pressure for 2 years. Clinical exam was unremarkable for syndromic features. There was no history of precocious puberty. Imaging studies confirm the presence of a large right adrenal mass with malignant features. Laboratory tests including the hormonal work up were unremarkable. Endocrinology team was involved in the perioperative management of the patient followed by a complete surgical excision of the tumor. Patient tolerated the procedure and discharged in good condition. Follow up appointments confirm the stabilization of the blood pressure and general condition with no evidence of recurrence at 1-year post-resection.
Adrenal tumors, Adrenocortical carcinoma, Abdominal mass, Secondary hypertension
Adrenocortical carcinomas (ACCs) are rare; the incidence is approximately one to two per million population per year as described by Ng L and Libertino JM [1]. They also emphasized that approximately 60 percent of ACCs are sufficiently secretory to present clinical syndrome of hormone excess [1]. Wajchenberg BL, et al. pointed that adults with hormone-secreting ACCs usually present with Cushing's syndrome alone (45%), or a mixed Cushing's and virilization syndrome, with overproduction of both glucocorticoids and androgens (25%) [2].
The clinical manifestations related to tumor growth (i.e., abdominal or flank pain) maybe the main presenting issues in most patients with nonfunctioning tumors (or more precisely with subclinical production of steroids), or with an incidentally found adrenal mass detected on radiographic imaging. The evaluation in apparently asymptomatic patients has been debated. Fassnacht M and Allolio B were involved in collaborative efforts (e.g. International Consensus Conference 2003 and networks like the European Network for the Study of Adrenal Tumours (ENSAT)) that recommended performing some tests to determine the secretory activity of the tumor even in asymptomatic patients [3]. Computed tomography (CT) scanning can usually distinguish adenomas from ACCs. Magnetic resonance imaging (MRI) is complementary to CT scan, in that local invasion and involvement of the surrounding major vasculature are more readily identifiable. The maximum diameter of the adrenal mass is predictive of malignancy. Most adrenal adenomas are less than 4 cm in diameter. In contrast, most ACCs are greater than 4 cm in diameter when discovered. Cytology from a specimen obtained by fine-needle aspiration (FNA) cannot distinguish a benign adrenal mass from adrenal carcinoma. It can, however, distinguish between an adrenal tumor and a metastatic tumor.
Pheochromocytoma should always be excluded by biochemical testing before attempting biopsy. Allolio B, et al. had concluded that complete surgical resection is the only potentially curative treatment for adrenocortical carcinoma (ACC) stage I-III [4]. Past results by Luton JP, et al. suggested a poor prognosis even for patients with early stage adrenal cancer [5]. However, some contemporary series studied by Icard P, et al. suggest that patients with ACC are living longer [6].
We report the case of a 19-year-old male referred to our center with a large adrenal mass. Past medical history was unremarkable until 2 years prior to presentation to outside institution when he was found to be hypertensive following a motor-vehicle accident, no detailed evaluation or report about the adrenal gland at that time. At the time, open fixation of a femoral fracture was delayed for blood pressure control. He was not prescribed anti-hypertensives on discharge. Social history included smoking and social drinking. More recently, he presented to a local Emergency Department with elevated blood pressure. He was prescribed angiotensin blockers and calcium channel blockers and referred to a tertiary center for work up of secondary hypertension given his young age. Further questioning revealed a one-year history of progressive episodes of intense attacks of palpitations accompanied by a sense of fear and anxiety, with elevated blood pressure readings during these episodes that became normal in between attacks. No history of weight gain change in appearance, muscle weakness or precocious puberty was given. Physical examination showed normal vital signs apart from slightly elevated blood pressure around 150/80 mmHg, weight 75 Kg, and height 184 cm. There were no clinical signs of Cushing's syndrome or virilization.
Laboratory work-up including full blood count, blood chemistry, serum electrolytes, liver function tests, cortisol, aldosterone, normetanephrines, total metanephrines, and urine metanephrines were all within normal ranges. Imaging showed a large right adrenal mass measuring 16 by 11 cm detected through CT and MRI of the abdomen (Figure 1a and Figure 1b). Despite negative biochemical and radiological findings, pre-operative probability of pheochromocytoma was thought to be high given the persistently elevated blood pressure. Therefore, the patient was started empirically on prazosin and later addition of propranolol.
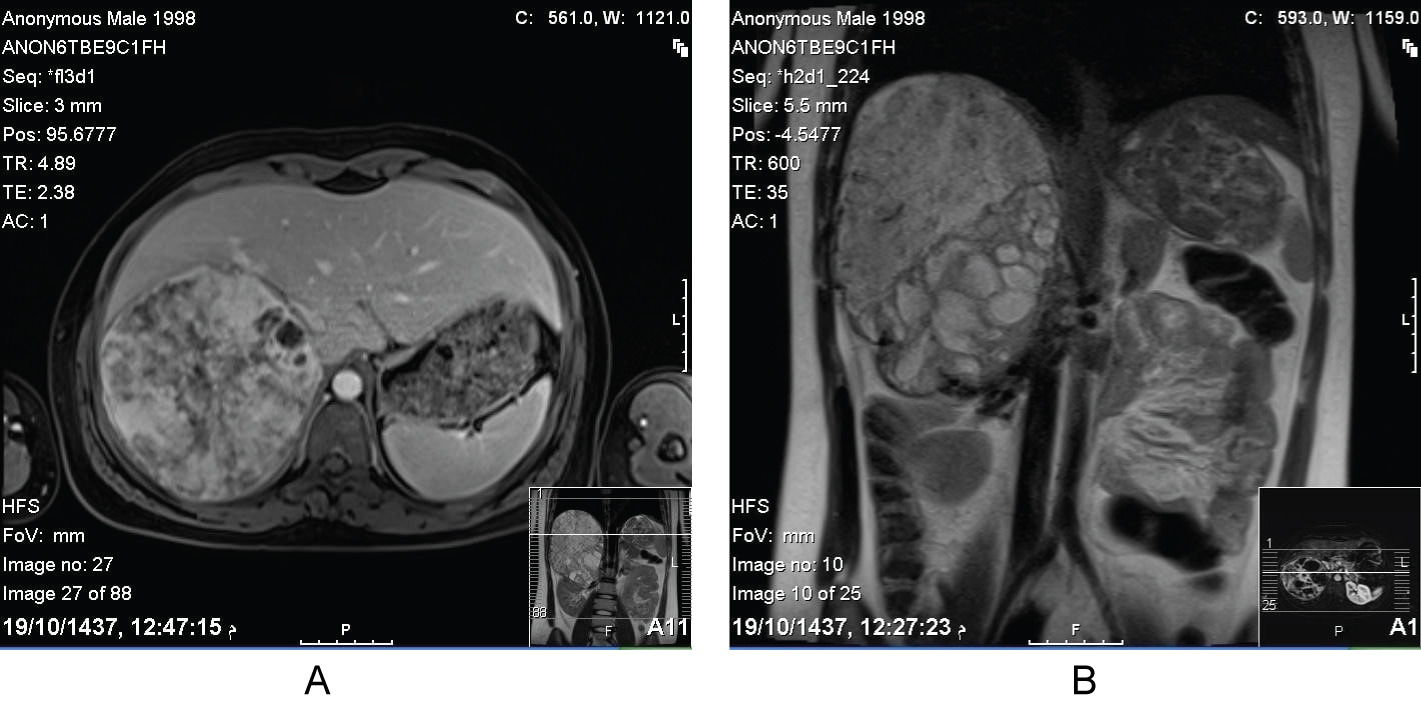 Figure 1: Axial and coronal sections of MRI abdomen showed the hetrogenous mass in the right adrenal gland.
View Figure 1
Figure 1: Axial and coronal sections of MRI abdomen showed the hetrogenous mass in the right adrenal gland.
View Figure 1
Open surgical approach was carried out. A 16 cm well-encapsulated mass was identified with dense adherence to the inferior vena cava and the posterior surface of the right liver. However, there was no evidence of gross invasion to the surrounding structure nor were there any obvious metastatic lesions. It was completely excised en-bloc with the right adrenal gland and the surrounding Gerota's fascia as well as the locoregional lymph nodes including the renal Hilary lymph nodes. The tumor capsule was not perforated and there was no need for vascular resection or frozen section biopsy. Blood pressure was monitored closely intra- and post-operatively without any incidence of increased blood pressure. The patient was released 5 days after surgery in good clinical condition.
The patient was followed up in the endocrinology clinic and the antihypertensive medications were weaned off by the first week post-operatively. Following discussion in the tumor board with the endocrinology and medical oncology, there was no indication to start adjuvant therapy as there was no evidence of positive tumor margin, lymphovascular invasion/involvement or distant metastases. In addition, the role of adjuvant therapy was felt of no benefit. The scheduled follow up was every 3 months for the first year then every 6 months thereafter till 5 years. So far, after 2 years of surgery, the patient did not show any evidence of local or distant recurrence radiologically. An informed consent was signed by the patient to report and publish his case.
MRI abdomen (Figure 1a and Figure 1b) showed a large right suprarenal mass measuring 16 × 8 × 11 cm. It demonstrated heterogenous predominantly bright T2 signal intensity and heterogenous slight T1 low signal intensity. The mass is displacing the surrounding organs and structures with no definite regional invasion. Malignant features are not clearly demonstrated. Primary vs. metastatic adrenal tumor vs hemorrhagic mass cannot be differentiated. Multiple lymph nodes are noted in the right renal fossa measuring approximately up to 1.1 cm. The visualized abdominal organs were otherwise unremarkable. No other special MRI techniques were used.
(Figure 2a and Figure 2b).
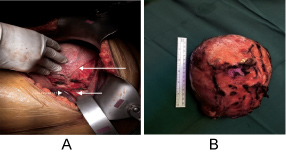 Figure 2: Intraoperative images show the huge (16 cm) adrenal mass (long arrow), the Inferior Vena Cava (short arrow) and the right liver lobe (dotted arrow).
View Figure 2
Figure 2: Intraoperative images show the huge (16 cm) adrenal mass (long arrow), the Inferior Vena Cava (short arrow) and the right liver lobe (dotted arrow).
View Figure 2
The slides submitted show adrenal gland with a large tumor; the specimen is reported to have measured 18 cm and weighed 590 g after formalin fixation. The tumor has variable architecture, composed mainly of nests and trabecula but also solid sheets of polygonal cells with eosinophilic cytoplasm and markedly pleomorphic nuclei. Mitoses are rare, and no atypical mitoses are identified. There is focal hemorrhage and necrosis. There is no frank capsular invasion in the slides examined, but there is unequivocal angioinvasion characterized by intravascular tumor cells associated with thrombus, both within the tumor and in the capsular vessels. The stroma shows edema, hyalinization, myxoid change and chondroid metaplasia with focal lipoblast-like features. The surrounding nontumorous adrenal cannot be evaluated.
The reticulin pattern is disrupted with areas of prominent pericellular deposition. The tumor cells exhibit strong nuclear reactivity for SF-1 but inhibin is only weakly and locally positive. They are positive for keratins using the AEI/AE3 cocktail, and exhibit reactivity for synaptophysin and vimentin, but they are negative for chromogranin and tyrosine hydroxylase. Staining for lGF-2 shows the granular perinuclear pattern characteristic of adrenal cortical carcinoma (Figure 3).
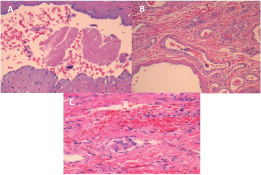 Figure 3: Some of the histological features of the resected tumor supporting the diagnosis of Adrenocortical Carcinoma.
View Figure 3
Figure 3: Some of the histological features of the resected tumor supporting the diagnosis of Adrenocortical Carcinoma.
View Figure 3
Beta-catenin shows nuclear translocation. Staining for p53 shows variable nuclear reactivity consistent with proliferating cells but not in the pattern indicative of mutation. 5100 highlights scattered cells in areas of chondroid metaplasia; SOX-10 is negative. Nuclear reactivity for MSH-2, MSN-6, MLH-1 and PMS-2 is intact. The Ki-67 labeling index is less than 2% in hot spots and the pHH3 stain confirms only rare mitotic figures.
The examined locoregional lymph nodes including the renal hilar lymph nodes were negative for tumor involvement.
Giving the unusual presentation of this case, the histopathological diagnosis was confirmed at 2 different institutions.
Adrenocortical carcinoma (ACC) is a rare neoplasm with poor prognosis. Nonfunctional variants of ACC were previously reported to be far less common than the functional types; The female-to-male ratio for ACCs is approximately 2.5-3:1. Male patients tend to be older and have a worse overall prognosis than do female patients. Although still somewhat controversial, Dehner LP, Hill DA suggest that children with ACC have a better prognosis than do adults; favorable clinical outcome has been reported in 70% or more of pediatric cases [7].
Unfortunately, most patients with adrenocortical carcinoma (ACC), specially the non-functional variant, present with advanced disease that is characterized by multiple abdominal or extra-abdominal metastatic masses (stage IV disease). Therefore, detection of tumors at an early clinical stage is crucial for curative resection; total resection offers the only prospect for cure. The National Italian Study Group review of adrenal incidentalomas summarized by Angeli A, et al. demonstrated that 90% of ACC cases had diameters of 4 cm or larger on radiologic imaging [8]. The estimated overall five-year survival rate for patients with ACC is approximately 20-35%. The experience of Lahey Clinic Medical center published by Tritos NA, et al. for cases in which total surgical resection is achieved, this rate is estimated to be approximately 32-47%, while in cases in which total surgical resection has not been possible, the five-year survival rate is estimated to be 10-30% [9]. As emphasized earlier, complete surgical resection remains the treatment of choice for the definitive management of ACC. The results of two different studies by Datrice NM, et al. and Hermsen IG concluded that even after apparently complete surgical resection, however, local or distant relapse occurs in nearly 80% of cases. In such settings, the only effective treatment is attempted reoperation [10,11]. As a long-term monitoring plan, monthly follow up sessions and scanning every 3 months for the first 2 years are the recommended strategy, based on limited evidence, after the surgical treatment of ACC because repeat resection for the recurrent disease can affect long-term survival.
Overall survival is poor for most patients with adrenocortical carcinoma. 45-60% is approximately the five-year survival for early stage disease and about 10-25% for advanced disease. Vassilopoulou-Sellin R and Schultz PN [12] had studied 139 adults treated for ACC over 20 years period at MD Anderson cancer center, 60% was the five-year survival rate, even though 34% had distant metastases at diagnosis.
After a careful searching in the literature, we found some similar reported cases of male and young (from late teens till mid-twenties) patient with huge (more than 10 cm in one dimension) mass of adrenocortical carcinoma (Table 1). However, there were limitations to access the details of some other non-English or historical articles. All those reported cases died shortly during the postoperative period. Although in our case, the patient is still in a good health condition and there was no evidence of recurrent or metastatic lesions during the first year follow up period after the challenging complete surgical resection.
Table 1: The reported cases of similar characteristics. View Table 1
We reported an extremely rare case of huge adrenocortical carcinoma in a young male patient who has a disease-free survival postoperative period. It is essential to differentiate it from an adrenocortical adenoma by correlating with clinical, biochemical, imaging, and histological features, because their prognoses are different. Complete surgical excision is the mainstay of treatment of adrenocortical carcinomas specially in early stages. Careful follow up plan should be established for such tumors with high recurrence rate.
There is no potential conflict of interest or funding agency involved in submission of this article.